

TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events
- PMID: 18838547
- PMCID: PMC2571935
- DOI: 10.1084/jem.20081370
TLR3 is an endogenous sensor of tissue necrosis during acute inflammatory events
Abstract
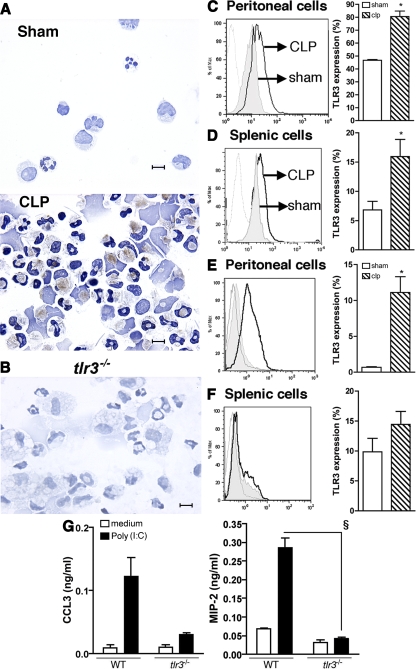
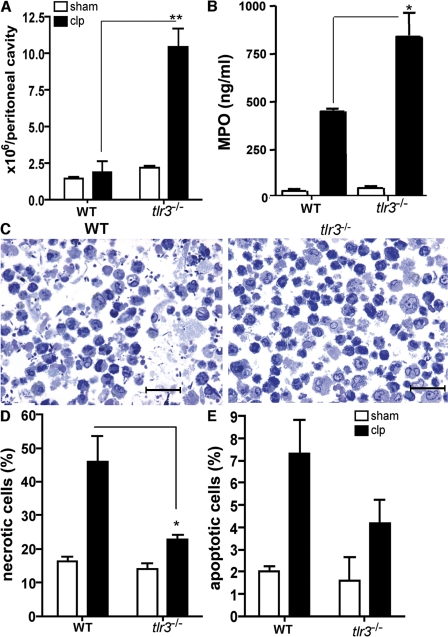
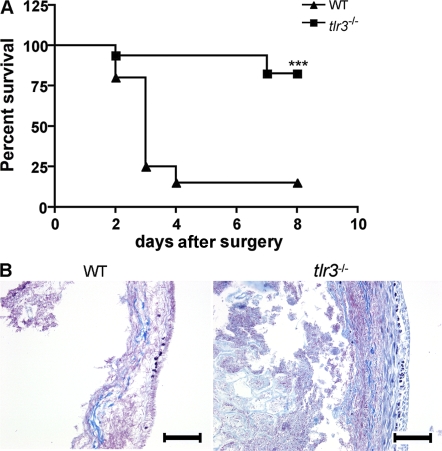
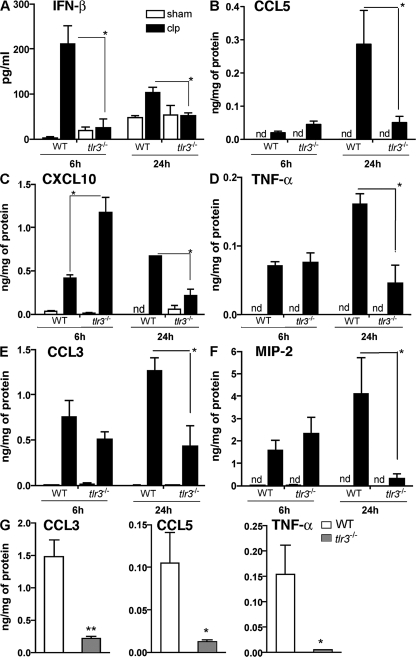
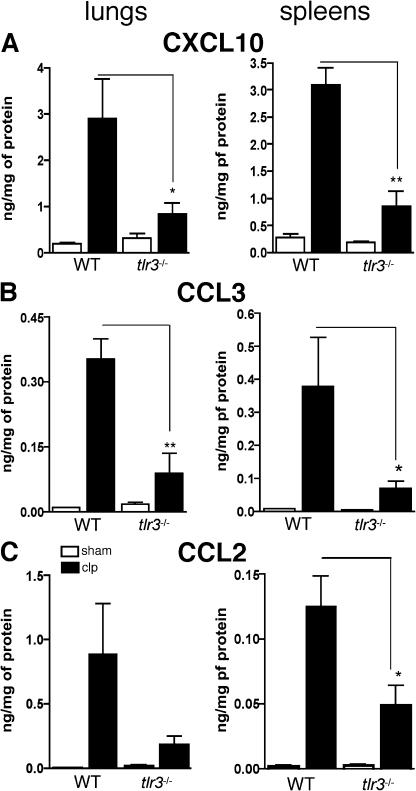
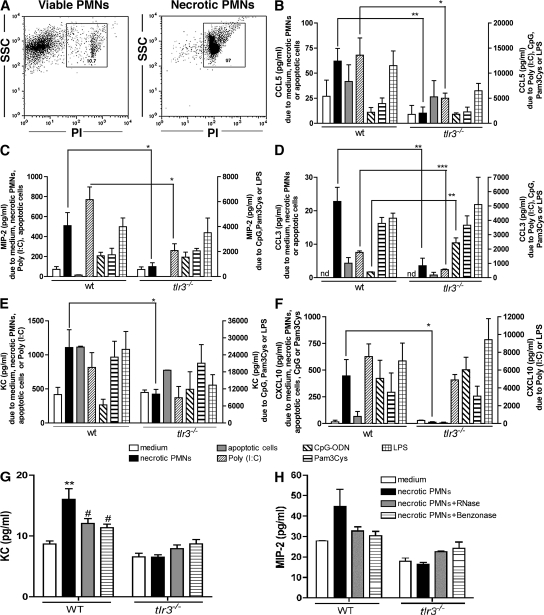
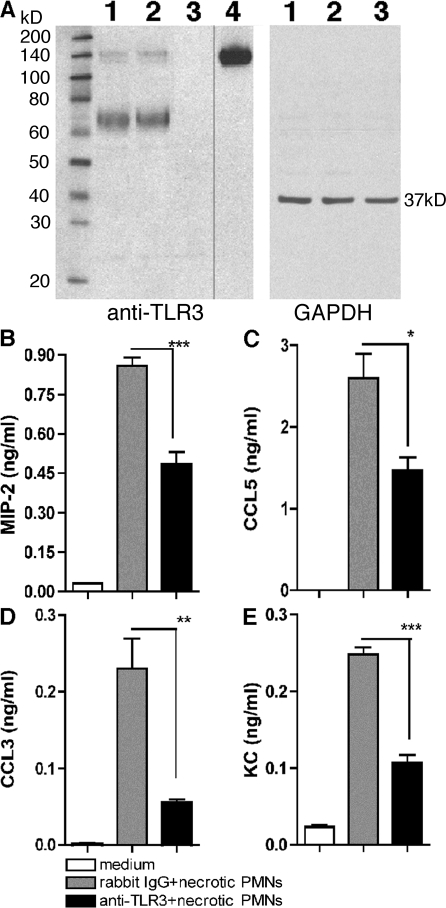
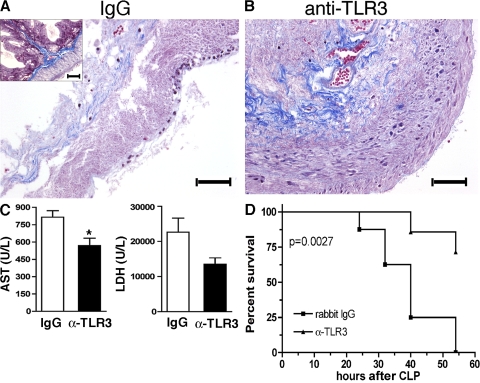
Ligands from dying cells are a source of Toll-like receptor (TLR) activating agents. Although TLR3 is known to respond to RNA from necrotic cells, the relative importance of this response in vivo during acute inflammatory processes has not been fully explored. We observed the involvement of TLR3 activation during experimental polymicrobial septic peritonitis and ischemic gut injury in the absence of an exogenous viral stimulus. In TLR3-deficient mice, increased chemokine/cytokine levels and neutrophil recruitment characterized the initial inflammatory responses in both injury models. However, the levels of inflammatory chemokines and tumor necrosis factor alpha quickly returned to baseline in tlr3(-/-) mice, and these mice were protected from the lethal effects of sustained inflammation. Macrophages from tlr3(-/-) mice responded normally to other TLR ligands but did not respond to RNA from necrotic neutrophils. Importantly, an immunoneutralizing antibody directed against TLR3 attenuated the generation of inflammatory chemokines evoked by byproducts from necrotic neutrophils cultured with wild-type macrophages. In vivo, anti-TLR3 antibody attenuated the tissue injury associated with gut ischemia and significantly decreased sepsis-induced mortality. Collectively, these data show that TLR3 is a regulator of the amplification of immune response and serves an endogenous sensor of necrosis, independent of viral activation.
Figures
References
-
- Pinsky, M.R. 2004. Dysregulation of the immune response in severe sepsis. Am. J. Med. Sci. 328:220–229. - PubMed
-
- Cohen, J. 2002. The immunopathogenesis of sepsis. Nature. 420:885–891. - PubMed
-
- Matzinger, P. 1994. Tolerance, danger, and the extended family. Annu. Rev. Immunol. 12:991–1045. - PubMed
-
- Gallucci, S., M. Lolkema, and P. Matzinger. 1999. Natural adjuvants: endogenous activators of dendritic cells. Nat. Med. 5:1249–1255. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
