

A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model
- PMID: 18838683
- PMCID: PMC2562418
- DOI: 10.1073/pnas.0807840105
A farnesyltransferase inhibitor prevents both the onset and late progression of cardiovascular disease in a progeria mouse model
Erratum in
- Proc Natl Acad Sci U S A. 2009 Aug 4;106(31):13143
Abstract
Hutchinson-Gilford progeria syndrome (HGPS) is the most dramatic form of human premature aging. Death occurs at a mean age of 13 years, usually from heart attack or stroke. Almost all cases of HGPS are caused by a de novo point mutation in the lamin A (LMNA) gene that results in production of a mutant lamin A protein termed progerin. This protein is permanently modified by a lipid farnesyl group, and acts as a dominant negative, disrupting nuclear structure. Treatment with farnesyltransferase inhibitors (FTIs) has been shown to prevent and even reverse this nuclear abnormality in cultured HGPS fibroblasts. We have previously created a mouse model of HGPS that shows progressive loss of vascular smooth muscle cells in the media of the large arteries, in a pattern that is strikingly similar to the cardiovascular disease seen in patients with HGPS. Here we show that the dose-dependent administration of the FTI tipifarnib (R115777, Zarnestra) to this HGPS mouse model can significantly prevent both the onset of the cardiovascular phenotype as well as the late progression of existing cardiovascular disease. These observations provide encouraging evidence for the current clinical trial of FTIs for this rare and devastating disease.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
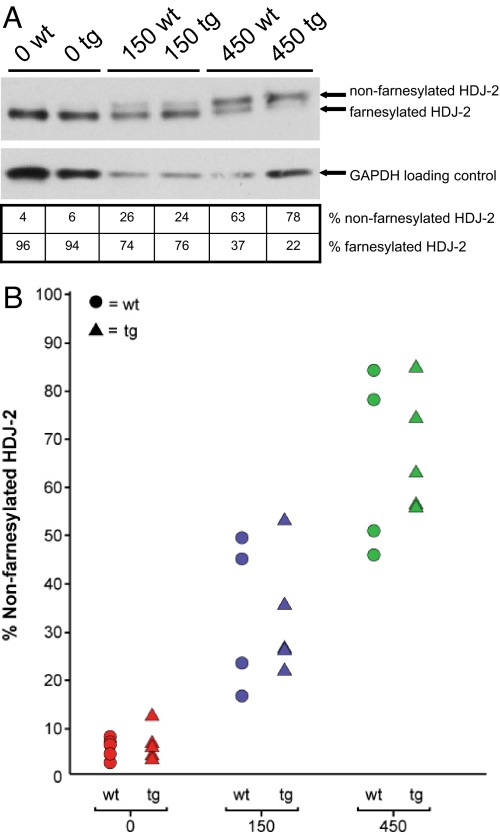
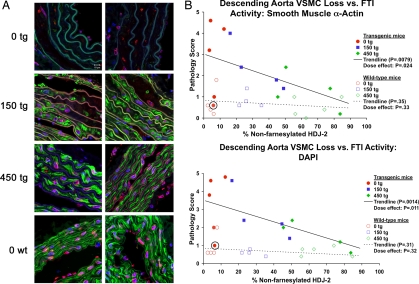
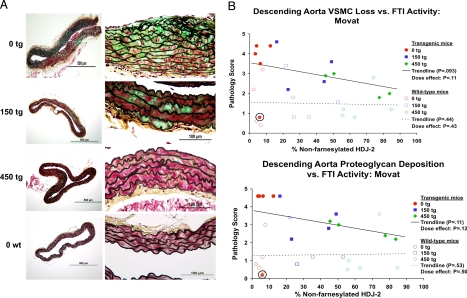
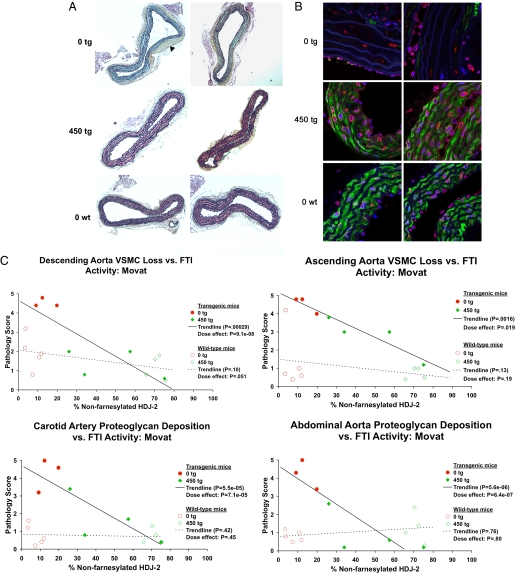
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
