

A database of phylogenetically atypical genes in archaeal and bacterial genomes, identified using the DarkHorse algorithm
- PMID: 18840280
- PMCID: PMC2573894
- DOI: 10.1186/1471-2105-9-419
A database of phylogenetically atypical genes in archaeal and bacterial genomes, identified using the DarkHorse algorithm
Abstract
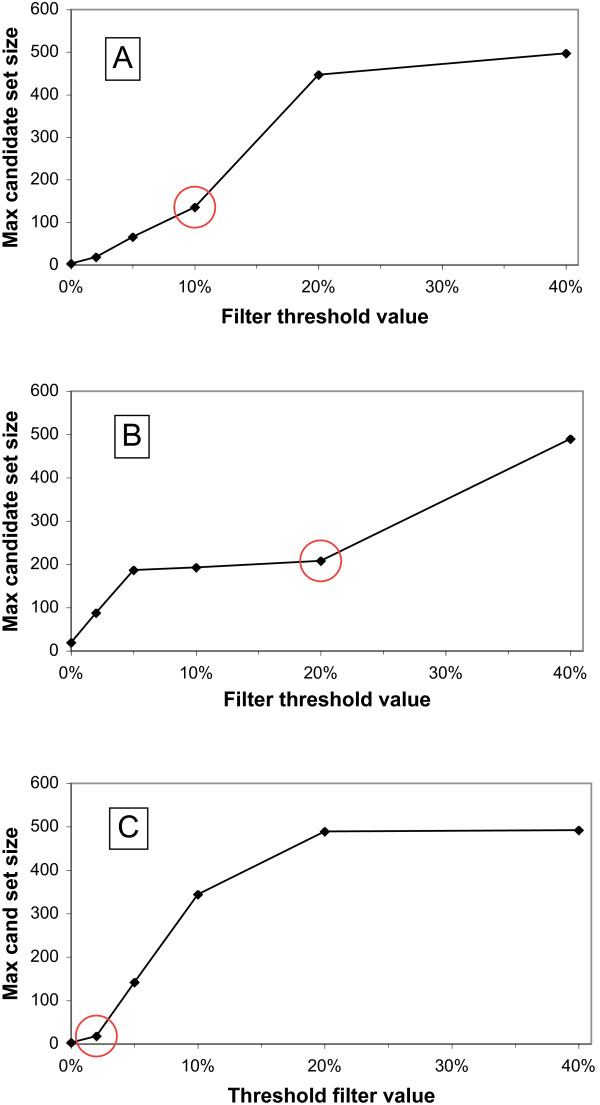
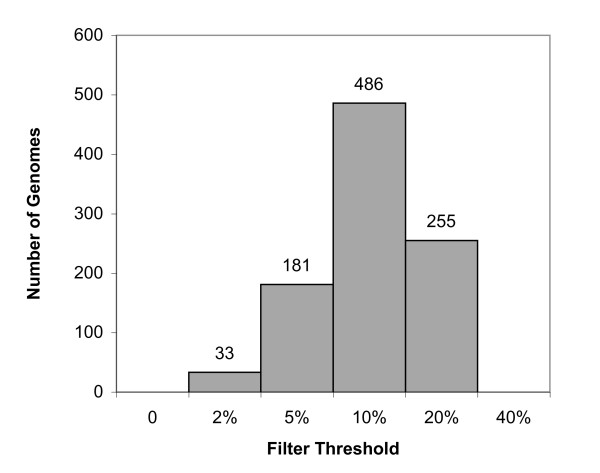
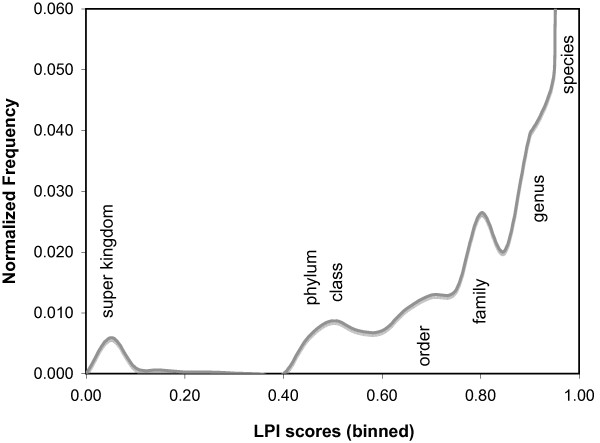
Background: The process of horizontal gene transfer (HGT) is believed to be widespread in Bacteria and Archaea, but little comparative data is available addressing its occurrence in complete microbial genomes. Collection of high-quality, automated HGT prediction data based on phylogenetic evidence has previously been impractical for large numbers of genomes at once, due to prohibitive computational demands. DarkHorse, a recently described statistical method for discovering phylogenetically atypical genes on a genome-wide basis, provides a means to solve this problem through lineage probability index (LPI) ranking scores. LPI scores inversely reflect phylogenetic distance between a test amino acid sequence and its closest available database matches. Proteins with low LPI scores are good horizontal gene transfer candidates; those with high scores are not.
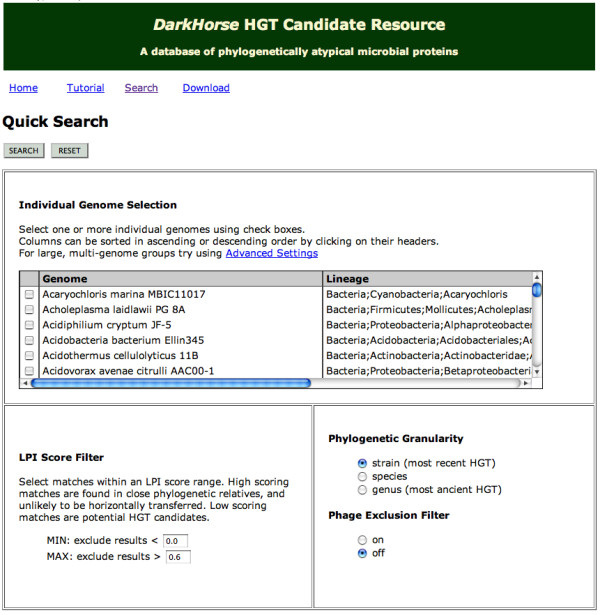
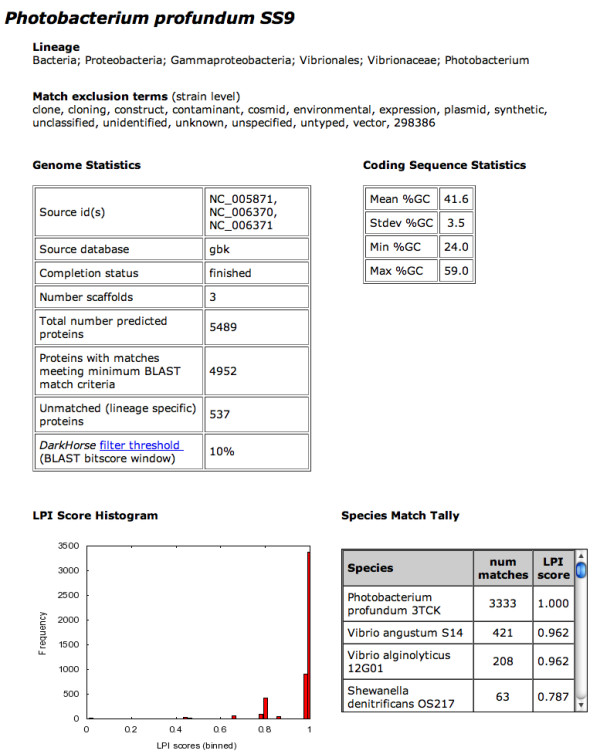
Description: The DarkHorse algorithm has been applied to 955 microbial genome sequences, and the results organized into a web-searchable relational database, called the DarkHorse HGT Candidate Resource http://darkhorse.ucsd.edu. Users can select individual genomes or groups of genomes to screen by LPI score, search for protein functions by descriptive annotation or amino acid sequence similarity, or select proteins with unusual G+C composition in their underlying coding sequences. The search engine reports LPI scores for match partners as well as query sequences, providing the opportunity to explore whether potential HGT donor sequences are phylogenetically typical or atypical within their own genomes. This information can be used to predict whether or not sufficient information is available to build a well-supported phylogenetic tree using the potential donor sequence.
Conclusion: The DarkHorse HGT Candidate database provides a powerful, flexible set of tools for identifying phylogenetically atypical proteins, allowing researchers to explore both individual HGT events in single genomes, and large-scale HGT patterns among protein families and genome groups. Although the DarkHorse algorithm cannot, by itself, provide definitive proof of horizontal gene transfer, it is a flexible, powerful tool that can be combined with slower, more rigorous methods in situations where these other methods could not otherwise be applied.
Figures

References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
