

p16INK4a-induced senescence is disabled by melanoma-associated mutations
- PMID: 18843795
- PMCID: PMC2582406
- DOI: 10.1111/j.1474-9726.2008.00422.x
p16INK4a-induced senescence is disabled by melanoma-associated mutations
Abstract
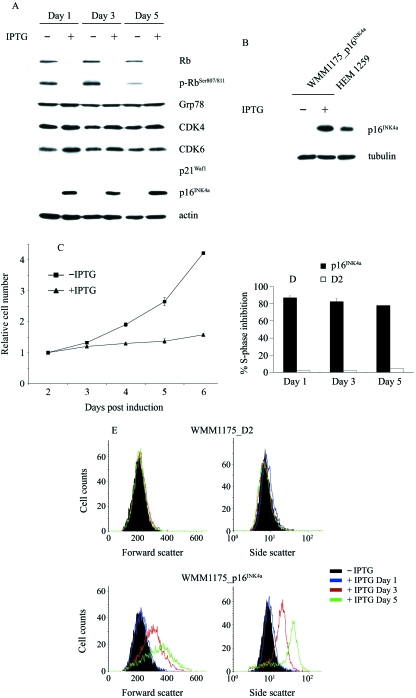
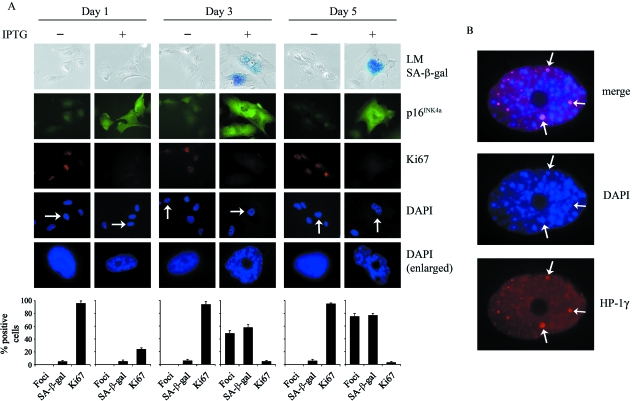
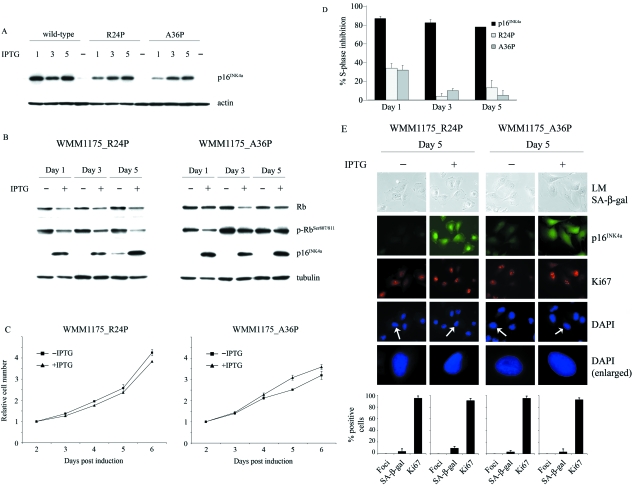
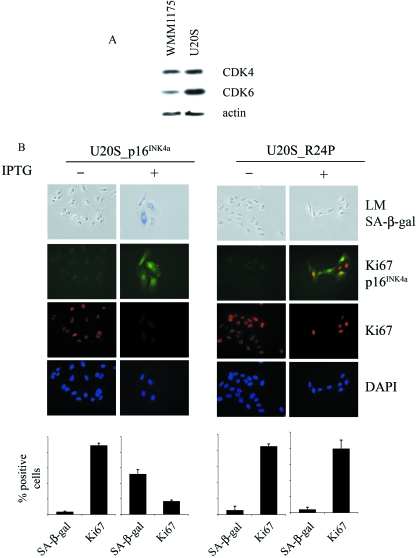
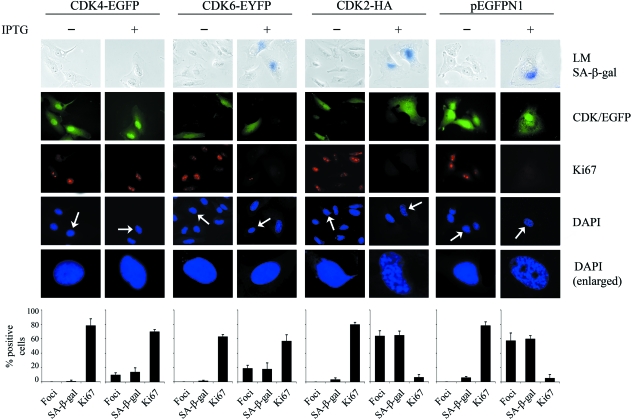
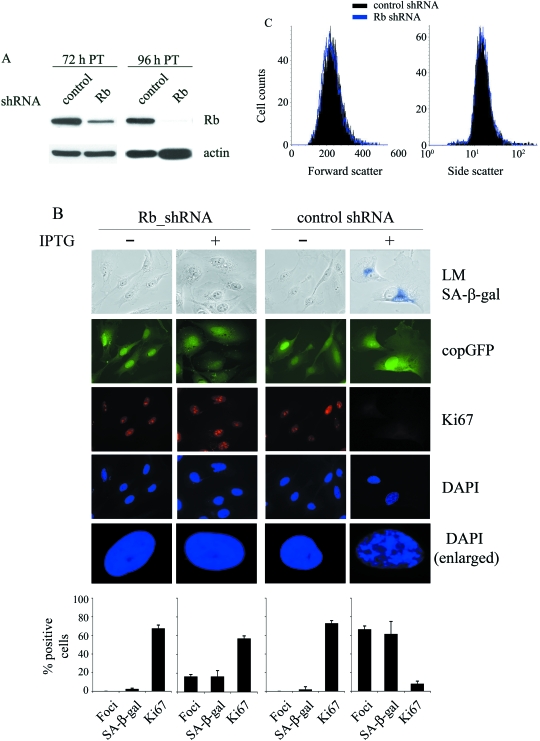
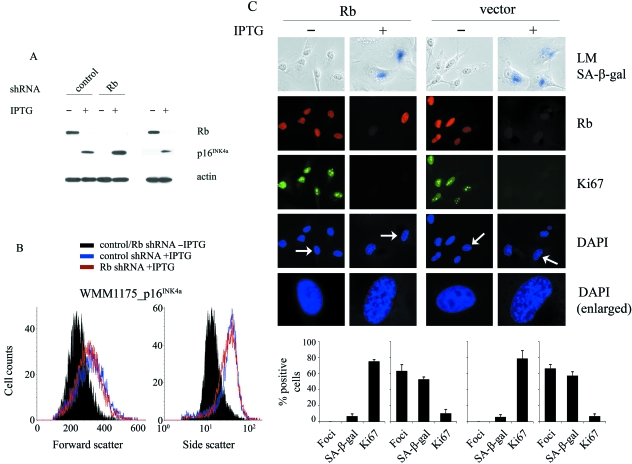
The p16(INK4a)-Rb tumour suppressor pathway is required for the initiation and maintenance of cellular senescence, a state of permanent growth arrest that acts as a natural barrier against cancer progression. Senescence can be overcome if the pathway is not fully engaged, and this may occur when p16(INK4a) is inactivated. p16(INK4a) is frequently altered in human cancer and germline mutations affecting p16(INK4a) have been linked to melanoma susceptibility. To characterize the functions of melanoma-associated p16(INK4a) mutations, in terms of promoting proliferative arrest and initiating senescence, we utilized an inducible expression system in a melanoma cell model. We show that wild-type p16(INK4a) promotes rapid cell cycle arrest that leads to a senescence programme characterized by the appearance of chromatin foci, activation of acidic beta-galactosidase activity, p53 independence and Rb dependence. Accumulation of wild-type p16(INK4a) also promoted cell enlargement and extensive vacuolization independent of Rb status. In contrast, the highly penetrant p16(INK4a) variants, R24P and A36P failed to arrest cell proliferation and did not initiate senescence. We also show that overexpression of CDK4, or its homologue CDK6, but not the downstream kinase, CDK2, inhibited the ability of wild-type p16(INK4a) to promote cell cycle arrest and senescence. Our data provide the first evidence that p16(INK4a) can initiate a CDK4/6-dependent autonomous senescence programme that is disabled by inherited melanoma-associated mutations.
Figures
References
-
- Ausserlechner MJ, Obexer P, Geley S, Kofler R. G1 arrest by p16 (INK4A) uncouples growth from cell cycle progression in leukemia cells with deregulated cyclin E and c-Myc expression. Leukemia. 2005;19:1051–1057. - PubMed
-
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, Takaoka M, Nakagawa H, Tort F, Fugger K, Johansson F, Sehested M, Andersen CL, Dyrskjot L, Orntoft T, Lukas J, Kittas C, Helleday T, Halazonetis TD, Bartek J, Gorgoulis VG. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature. 2006;444:633–637. - PubMed
-
- Becker TM, Rizos H, Kefford RF, Mann GJ. Functional impairment of melanoma-associated p16INK4a mutants in melanoma cells despite retention of cyclin-dependent kinase 4 binding. Clin. Cancer Res. 2001;7:3282–3288. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
