

Elevated synaptic activity preconditions neurons against an in vitro model of ischemia
- PMID: 18845540
- PMCID: PMC3259903
- DOI: 10.1074/jbc.M805624200
Elevated synaptic activity preconditions neurons against an in vitro model of ischemia
Abstract
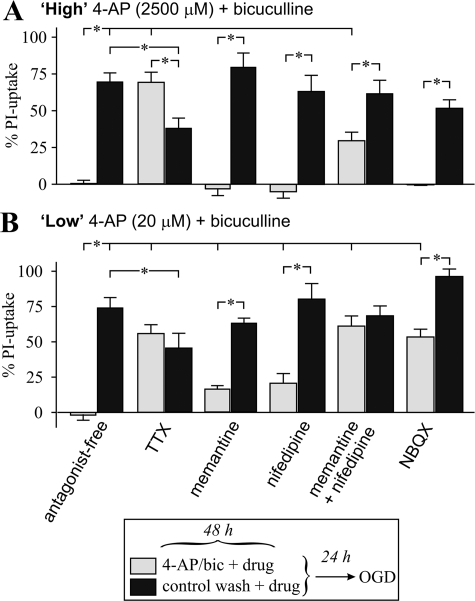
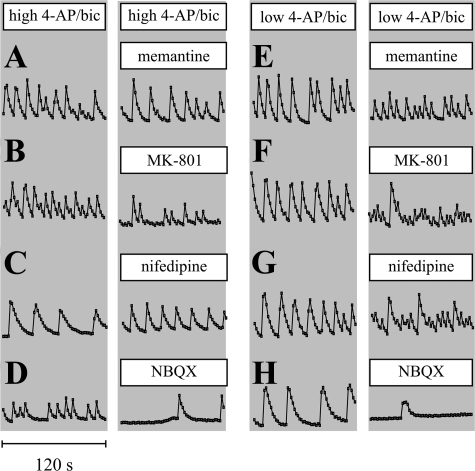
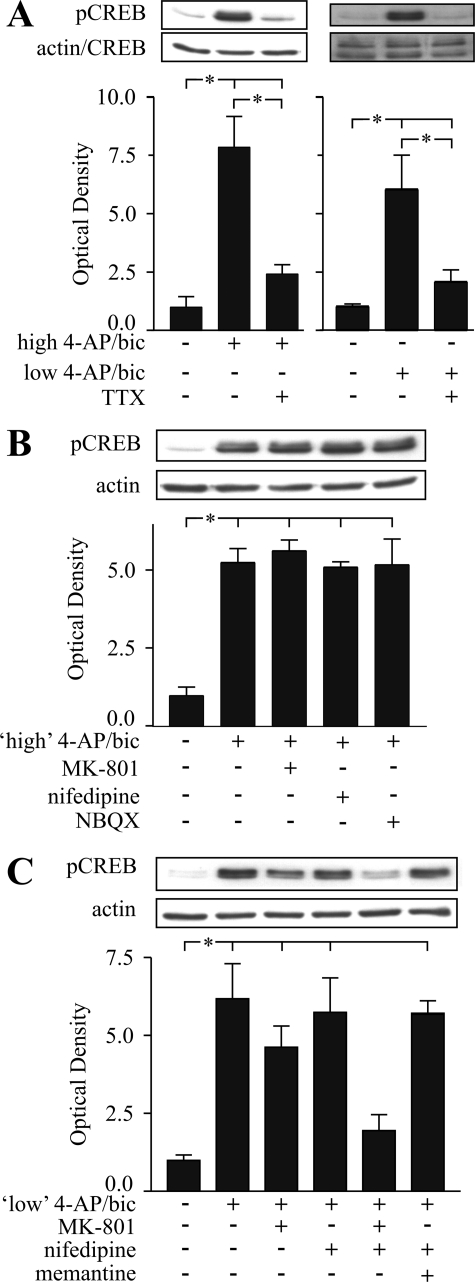
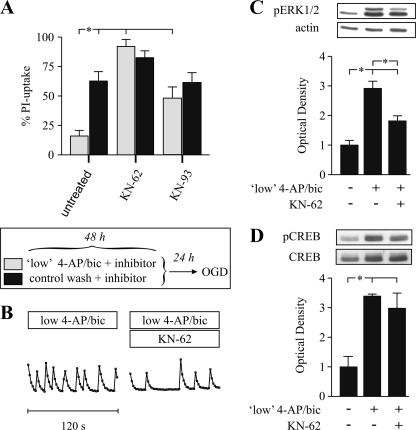
Tolerance to otherwise lethal cerebral ischemia in vivo or to oxygen-glucose deprivation (OGD) in vitro can be induced by prior transient exposure to N-methyl-D-aspartic acid (NMDA): preconditioning in this manner activates extrasynaptic and synaptic NMDA receptors and can require bringing neurons to the "brink of death." We considered if this stressful requirement could be minimized by the stimulation of primarily synaptic NMDA receptors. Subjecting cultured cortical neurons to prolonged elevations in electrical activity induced tolerance to OGD. Specifically, exposing cultures to a K(+)-channel blocker, 4-aminopyridine (20-2500 microm), and a GABA(A) receptor antagonist, bicuculline (50 microm) (4-AP/bic), for 1-2 days resulted in potent tolerance to normally lethal OGD applied up to 3 days later. Preconditioning induced phosphorylation of ERK1/2 and CREB which, along with Ca(2+) spiking and OGD tolerance, was eliminated by tetrodotoxin. Antagonists of NMDA receptors or L-type voltage-gated Ca(2+) channels (L-VGCCs) applied during preconditioning decreased Ca(2+) spiking, phosphorylation of ERK1/2 and CREB, and OGD tolerance more effectively when combined, particularly at the lowest 4-AP concentration. Inhibiting ERK1/2 or Ca(2+)/calmodulin-dependent protein kinases (CaMKs) also reduced Ca(2+) spiking and OGD tolerance. Preconditioning resulted in altered neuronal excitability for up to 3 days following 4-AP/bic washout, based on field potential recordings obtained from neurons cultured on 64-channel multielectrode arrays. Taken together, the data are consistent with action potential-driven co-activation of primarily synaptic NMDA receptors and L-VGCCs, resulting in parallel phosphorylation of ERK1/2 and CREB and involvement of CaMKs, culminating in a potent, prolonged but reversible, OGD-tolerant phenotype.
Figures


References
-
- Kirino, T. (2002) J. Cereb. Blood Flow Metab. 22 1283–1296 - PubMed
-
- Tauskela, J. S., Gendron, T., and Morley, P. (2004) in: Cerebral Ischemic Tolerance (Schaller, B., ed) pp. 45–94, Nova Science Publishers, Inc., Hauppauge
-
- Kato, H., Liu, Y., Araki, T., and Kogure, K. (1992) Neurosci. Lett. 139 118–121 - PubMed
-
- Bond, A., Lodge, D., Hicks, C. A., Ward, M. A., and O'Neill, M. J. (1999) Eur. J. Pharmacol. 380 91–99 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
