

Protein kinase D1 mediates NF-kappaB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells
- PMID: 18845574
- PMCID: PMC2604803
- DOI: 10.1152/ajpgi.90452.2008
Protein kinase D1 mediates NF-kappaB activation induced by cholecystokinin and cholinergic signaling in pancreatic acinar cells
Abstract
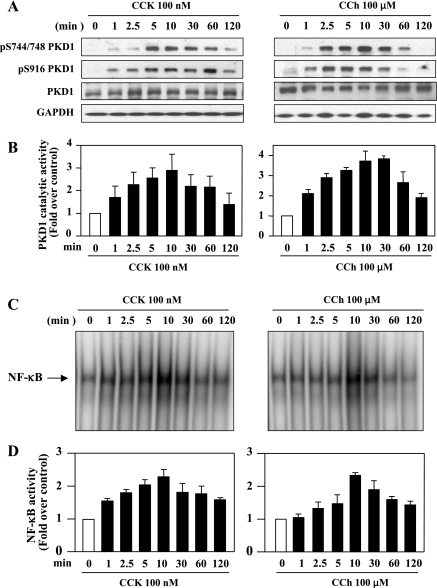
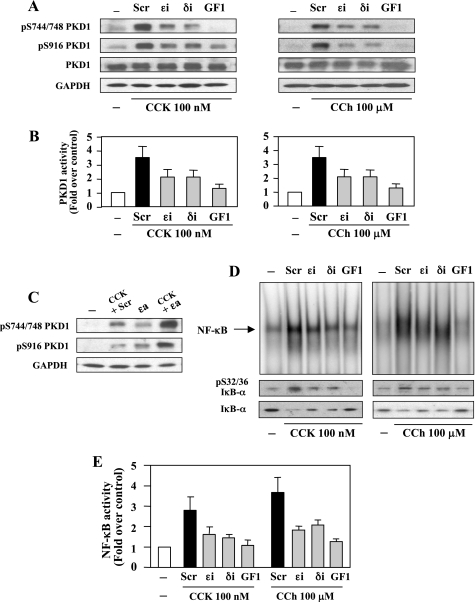
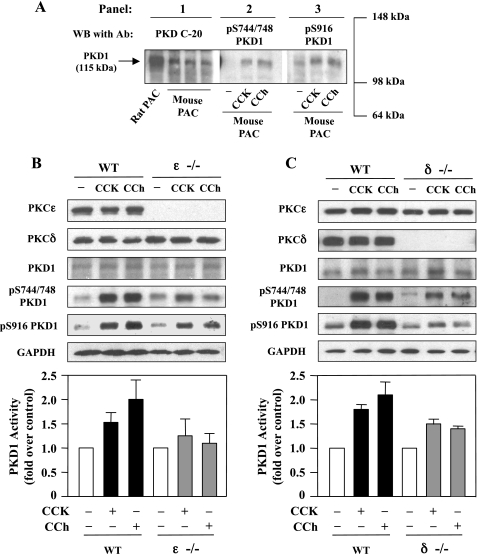
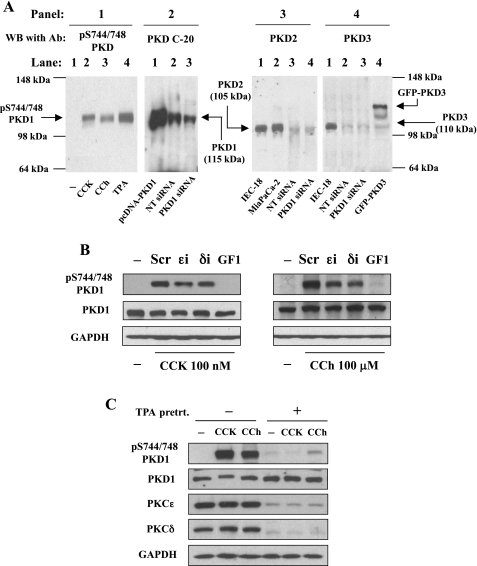
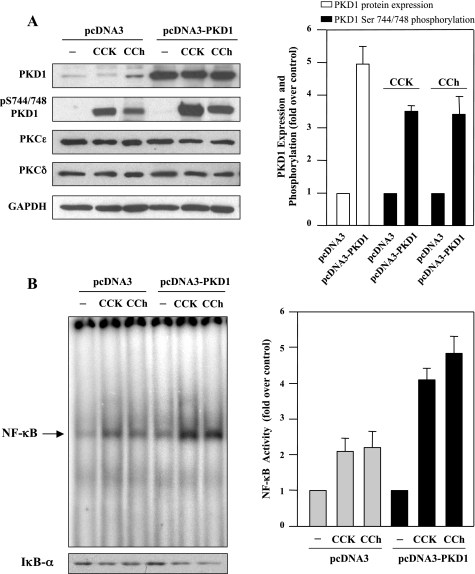
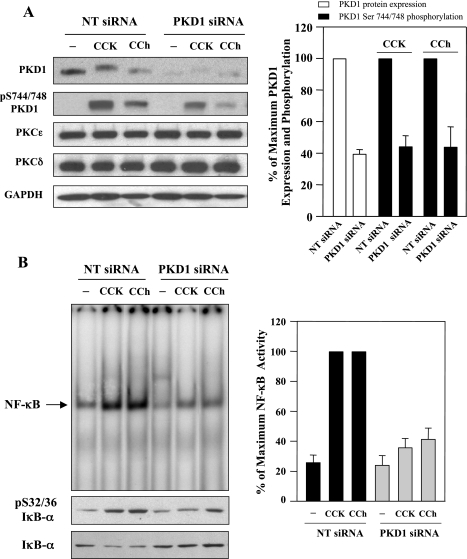
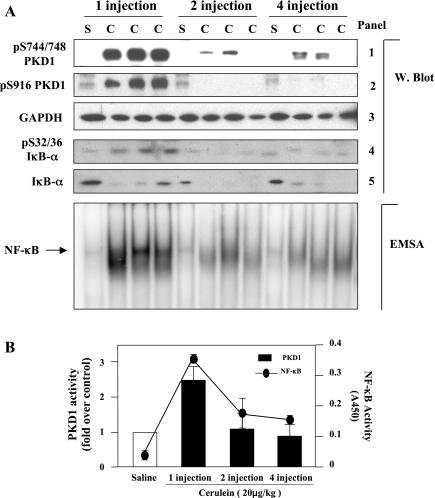
The transcription factor NF-kappaB plays a critical role in inflammatory and cell death responses during acute pancreatitis. Previous studies in our laboratory demonstrated that protein kinase C (PKC) isoforms PKCdelta and epsilon are key regulators of NF-kappaB activation induced by cholecystokinin-8 (CCK-8), tumor necrosis factor-alpha, and ethanol. However, the downstream participants in regulating NF-kappaB activation in exocrine pancreas remain poorly understood. Here, we demonstrate that protein kinase D1 (PKD1) is a key downstream target of PKCdelta and PKCepsilon in pancreatic acinar cells stimulated by two major secretagogues, CCK-8 and the cholinergic agonist carbachol (CCh), and that PKD1 is necessary for NF-kappaB activation induced by CCK-8 and CCh. Both CCK-8 and CCh dose dependently induced a rapid and striking activation of PKD1 in rat pancreatic acinar cells, as measured by in vitro kinase assay and by phosphorylation at PKD1 activation loop (Ser744/748) or autophosphorylation site (Ser916). The phosphorylation and activation of PKD1 correlated with NF-kappaB activity stimulated by CCK-8 or CCh, as measured by NF-kappaB DNA binding. Either inhibition of PKCdelta or epsilon by isoform-specific inhibitory peptides, genetic deletion of PKCdelta and epsilon in pancreatic acinar cells, or knockdown of PKD1 by using small interfering RNAs in AR42J cells resulted in a marked decrease in PKD1 and NF-kappaB activation stimulated by CCK-8 or CCh. Conversely, overexpression of PKD1 resulted in augmentation of CCK-8- and CCh-stimulated NF-kappaB activation. Finally, the kinetics of PKD1 and NF-kappaB activation during cerulein-induced rat pancreatitis showed that both PKD1 and NF-kappaB activation were early events during acute pancreatitis and that their time courses of response were similar. Our results identify PKD1 as a novel early convergent point for PKCdelta and epsilon in the signaling pathways mediating NF-kappaB activation in pancreatitis.
Figures
References
-
- Adler G, Beglinger C, Braun U, Reinshagen M, Koop I, Schafmayer A, Rovati L, Arnold R. Interaction of the cholinergic system and cholecystokinin in the regulation of endogenous and exogenous stimulation of pancreatic secretion in humans. Gastroenterology 100: 537–543, 1991. - PubMed
-
- Akiyama T, Hirohata Y, Okabayashi Y, Imoto I, Otsuki M. Supramaximal CCK and CCh concentrations abolish VIP potentiation by inhibiting adenylyl cyclase activity. Am J Physiol Gastrointest Liver Physiol 275: G1202–G1208, 1998. - PubMed
-
- Bastani B, Yang L, Baldassare JJ, Pollo DA, Gardner JD. Cellular distribution of isoforms of protein kinase C (PKC) in pancreatic acini. Biochim Biophys Acta 1269: 307–315, 1995. - PubMed
-
- Chen X, Ji B, Han B, Ernst SA, Simeone D, Logsdon CD. NF-kappaB activation in pancreas induces pancreatic and systemic inflammatory response. Gastroenterology 122: 448–457, 2002. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases