

Endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes is opposed by compensatory overexpression of cyclooxygenase-2 in the vasculature
- PMID: 18845644
- PMCID: PMC2646543
- DOI: 10.1210/en.2008-1069
Endothelial dysfunction in mice with streptozotocin-induced type 1 diabetes is opposed by compensatory overexpression of cyclooxygenase-2 in the vasculature
Abstract
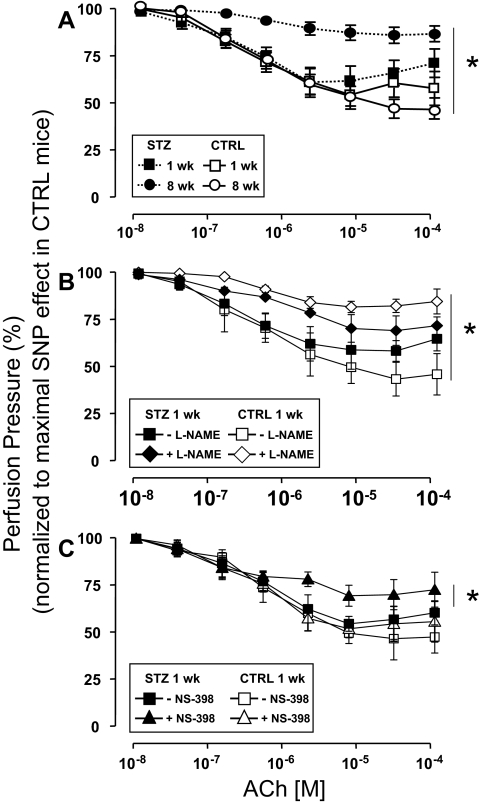
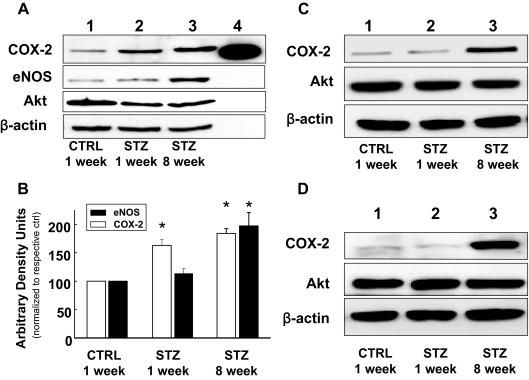
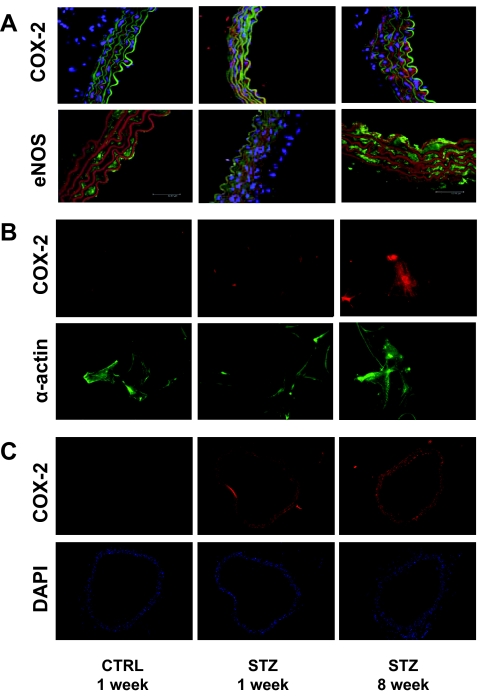
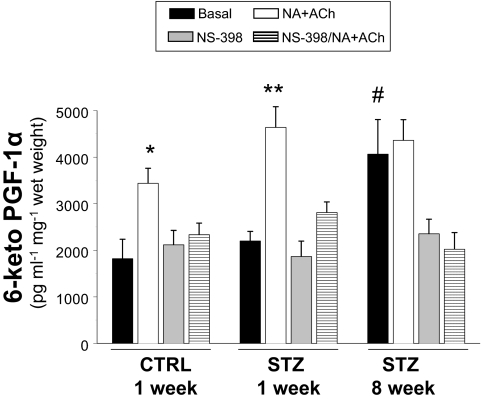
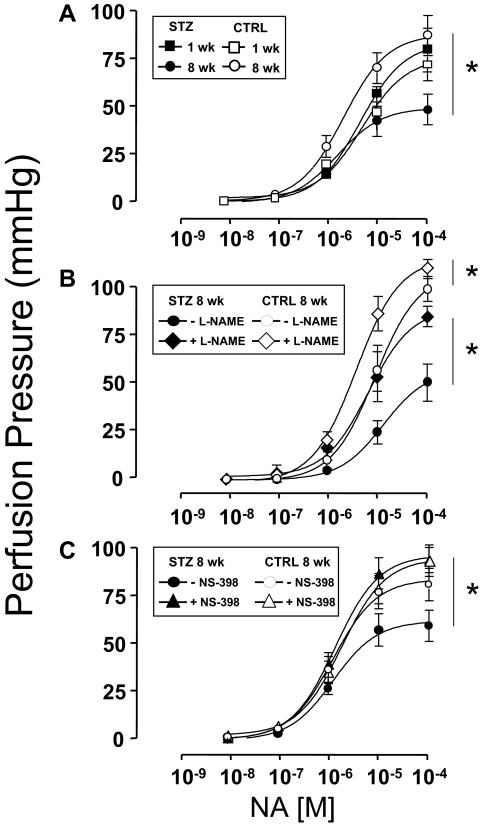
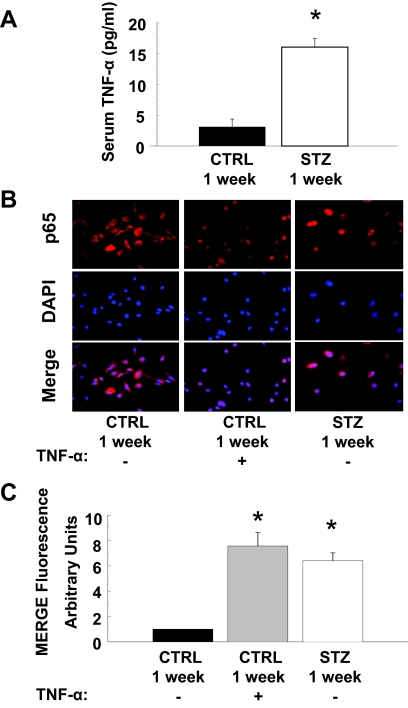
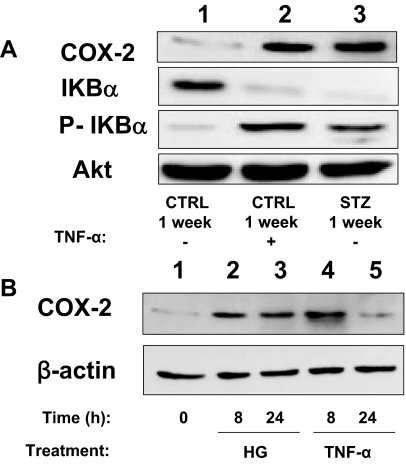
Cardiovascular complications of diabetes result from endothelial dysfunction secondary to persistent hyperglycemia. We investigated potential compensatory mechanisms in the vasculature that oppose endothelial dysfunction in diabetes. BALB/c mice were treated with streptozotocin (STZ) to induce type 1 diabetes (T1D). In mesenteric vascular beds (MVBs), isolated ex vivo from mice treated with STZ for 1 wk, dose-dependent vasorelaxation to acetylcholine (ACh) or sodium nitroprusside was comparable with that in age-matched control mice (CTRL). By contrast, MVBs from mice treated with STZ for 8 wk had severely impaired vasodilator responses to ACh consistent with endothelial dysfunction. Pretreatment of MVBs from CTRL mice with nitric oxide synthase inhibitor nearly abolished vasodilation to ACh. In MVB from 1-wk STZ-treated mice, vasodilation to ACh was only partially impaired by L-N(omega)-arginine methyl ester. Thus, vasculature of mice with T1D may have compensatory nitric oxide-independent mechanisms to augment vasodilation to ACh and oppose endothelial dysfunction. Indeed, pretreatment of MVBs isolated from 1-wk STZ-treated mice with NS-398 [selective cyclooxygenase (COX)-2 inhibitor] unmasked endothelial dysfunction not evident in CTRL mice pretreated without or with NS-398. Expression of COX-2 in MVBs, aortic endothelial cells, and aortic vascular smooth muscle cells from STZ-treated mice was significantly increased (vs. CTRL). Moreover, concentrations of the COX-2-dependent vasodilator 6-keto-prostaglandin F-1alpha was elevated in conditioned media from aorta of STZ-treated mice. We conclude that endothelial dysfunction in a mouse model of T1D is opposed by compensatory up-regulation of COX-2 expression and activity in the vasculature that may be relevant to developing novel therapeutic strategies for diabetes and its cardiovascular complications.
Figures
References
-
- Bianchi C, Miccoli R, Penno G, Del Prato S 2008 Primary prevention of cardiovascular disease in people with dysglycemia. Diabetes Care 31(Suppl 2):S208–S214 - PubMed
-
- Muniyappa R, Montagnani M, Koh KK, Quon MJ 2007 Cardiovascular actions of insulin. Endocr Rev 28:463–491 - PubMed
-
- Kim JA, Montagnani M, Koh KK, Quon MJ 2006 Reciprocal relationships between insulin resistance and endothelial dysfunction: molecular and pathophysiological mechanisms. Circulation 113:1888–1904 - PubMed
-
- Sheetz MJ, King GL 2002 Molecular understanding of hyperglycemia’s adverse effects for diabetic complications. JAMA 288:2579–2588 - PubMed
-
- Deanfield JE, Halcox JP, Rabelink TJ 2007 Endothelial function and dysfunction: testing and clinical relevance. Circulation 115:1285–1295 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
