

High-resolution mapping of expression-QTLs yields insight into human gene regulation
- PMID: 18846210
- PMCID: PMC2556086
- DOI: 10.1371/journal.pgen.1000214
High-resolution mapping of expression-QTLs yields insight into human gene regulation
Abstract
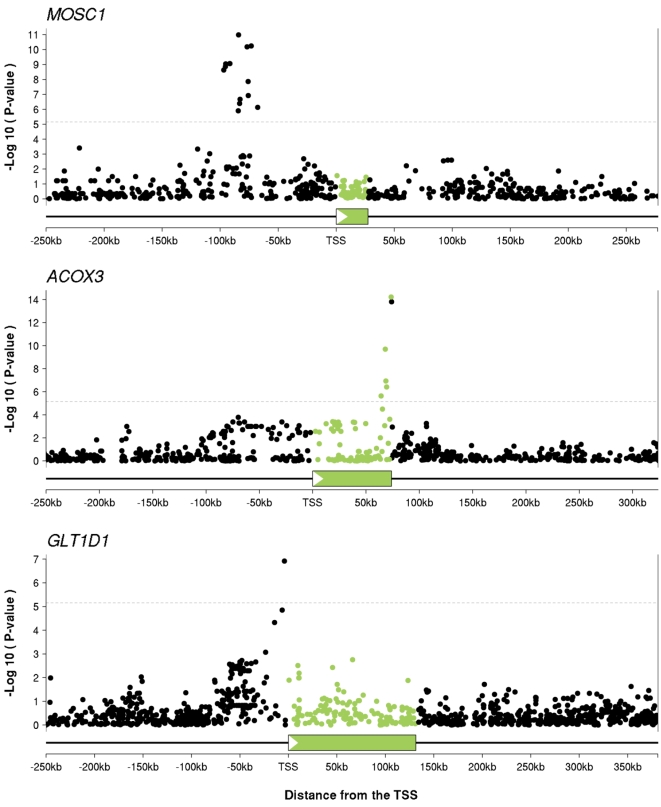
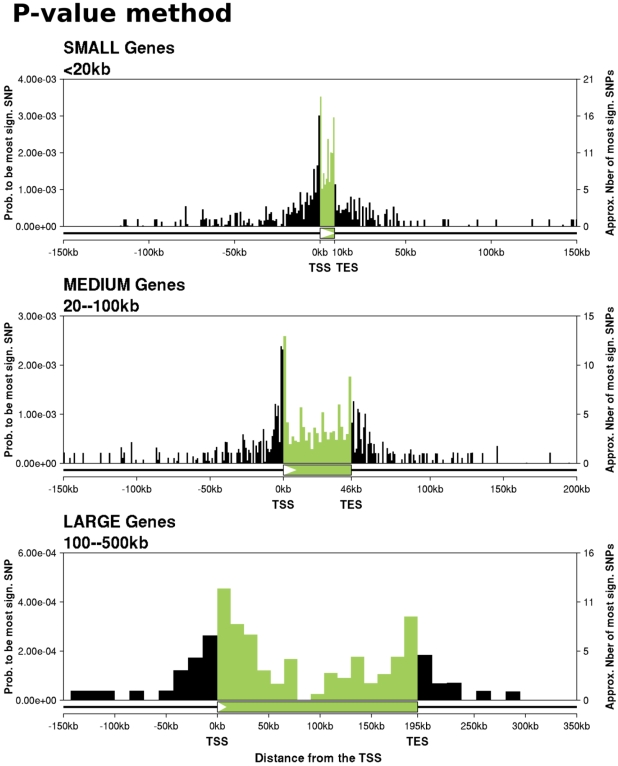
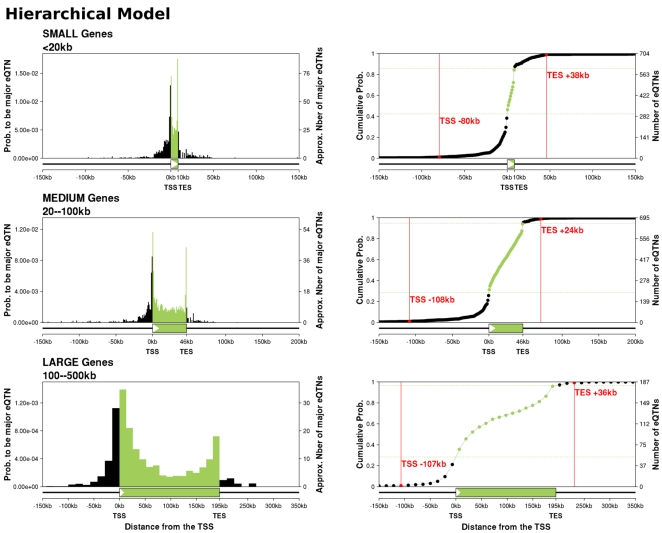
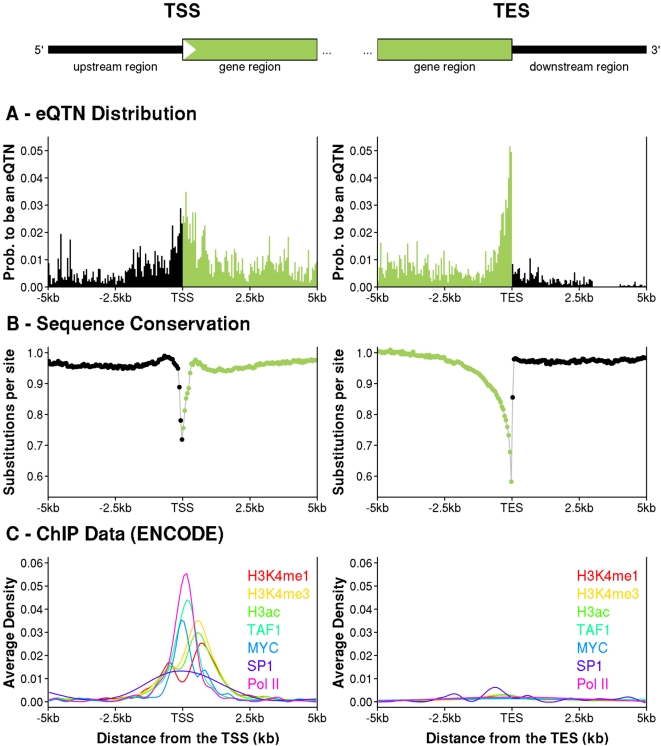
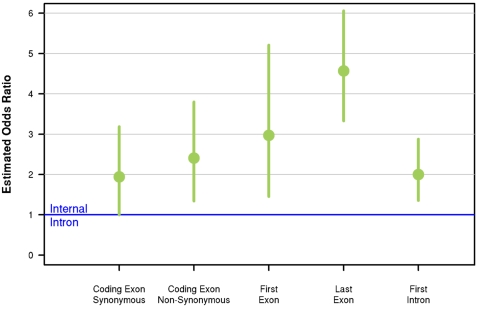
Recent studies of the HapMap lymphoblastoid cell lines have identified large numbers of quantitative trait loci for gene expression (eQTLs). Reanalyzing these data using a novel Bayesian hierarchical model, we were able to create a surprisingly high-resolution map of the typical locations of sites that affect mRNA levels in cis. Strikingly, we found a strong enrichment of eQTLs in the 250 bp just upstream of the transcription end site (TES), in addition to an enrichment around the transcription start site (TSS). Most eQTLs lie either within genes or close to genes; for example, we estimate that only 5% of eQTLs lie more than 20 kb upstream of the TSS. After controlling for position effects, SNPs in exons are approximately 2-fold more likely than SNPs in introns to be eQTLs. Our results suggest an important role for mRNA stability in determining steady-state mRNA levels, and highlight the potential of eQTL mapping as a high-resolution tool for studying the determinants of gene regulation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Wray G. The evolutionary significance of cis-regulatory mutations. Nat Rev Genet. 2007;8:206–216. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
