

Review. The origin of the prion agent of kuru: molecular and biological strain typing
- PMID: 18849291
- PMCID: PMC2581656
- DOI: 10.1098/rstb.2008.0069
Review. The origin of the prion agent of kuru: molecular and biological strain typing
Abstract
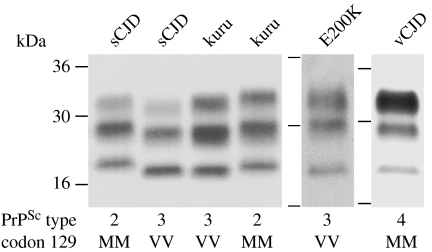
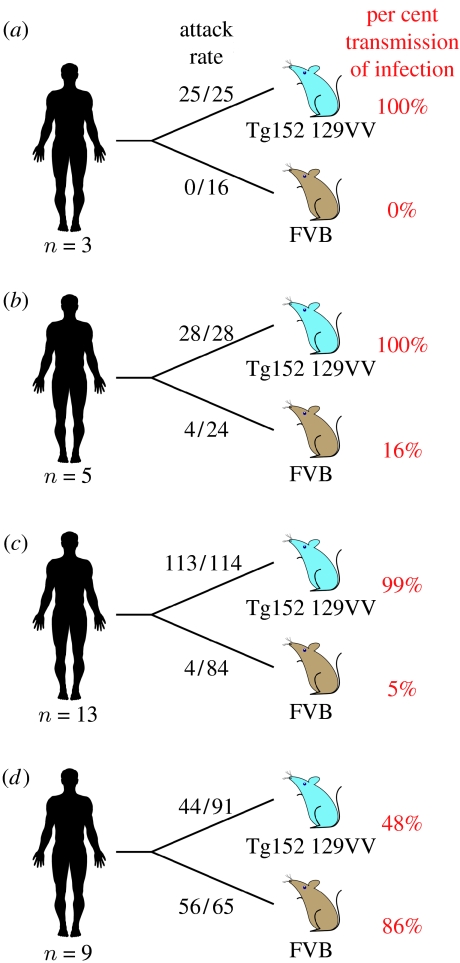
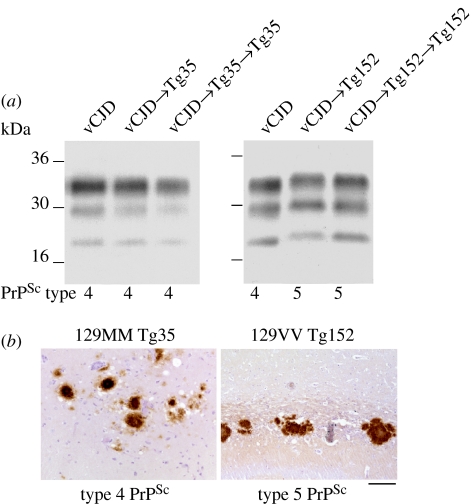
Kuru is an acquired human prion disease that primarily affected the Fore linguistic group of the Eastern Highlands of Papua New Guinea. The central clinical feature of kuru is progressive cerebellar ataxia and, in sharp contrast to most cases of sporadic Creutzfeldt-Jakob disease (CJD), dementia is a less prominent and usually late clinical feature. In this regard, kuru is more similar to variant CJD, which also has similar prodromal symptoms of sensory disturbance and joint pains in the legs and psychiatric and behavioural changes. Since a significant part of the clinicopathological diversity seen in human prion disease is likely to relate to the propagation of distinct human prion strains, we have compared the transmission properties of kuru prions with those isolated from patients with sporadic, iatrogenic and variant CJD in both transgenic and wild-type mice. These data have established that kuru prions have prion strain properties equivalent to those of classical (sporadic and iatrogenic) CJD prions but distinct from variant CJD prions. Here, we review these findings and discuss how peripheral routes of infection and other factors may be critical modifiers of the kuru phenotype.
Figures

References
-
- Alpers M. Epidemiology and clinical aspects of kuru. In: Prusiner S.B., McKinley M.P., editors. Prions: novel infectious pathogens causing scrapie and Creutzfeldt–Jakob disease. Academic Press; San Diego, CA: 1987. pp. 451–465.
-
- Alpers M., Rail L. Kuru and Creutzfeldt–Jakob disease: clinical and aetiological aspects. Proc. Aust. Assoc. Neurol. 1971;8:7–15. - PubMed
-
- Asante E.A., et al. BSE prions propagate as either variant CJD-like or sporadic CJD-like prion strains in transgenic mice expressing human prion protein. EMBO J. 2002;21:6358–6366. doi:10.1093/emboj/cdf653 - DOI - PMC - PubMed
-
- Asante E.A., et al. Dissociation of pathological and molecular phenotype of variant Creutzfeldt–Jakob disease in transgenic human prion protein 129 heterozygous mice. Proc. Natl Acad. Sci. USA. 2006;103:10 759–10 764. doi:10.1073/pnas.0604292103 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical