

Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases
- PMID: 18849971
- PMCID: PMC2880455
- DOI: 10.1038/nchembio.117
Targeted polypharmacology: discovery of dual inhibitors of tyrosine and phosphoinositide kinases
Abstract
The clinical success of multitargeted kinase inhibitors has stimulated efforts to identify promiscuous drugs with optimal selectivity profiles. It remains unclear to what extent such drugs can be rationally designed, particularly for combinations of targets that are structurally divergent. Here we report the systematic discovery of molecules that potently inhibit both tyrosine kinases and phosphatidylinositol-3-OH kinases, two protein families that are among the most intensely pursued cancer drug targets. Through iterative chemical synthesis, X-ray crystallography and kinome-level biochemical profiling, we identified compounds that inhibit a spectrum of new target combinations in these two families. Crystal structures revealed that the dual selectivity of these molecules is controlled by a hydrophobic pocket conserved in both enzyme classes and accessible through a rotatable bond in the drug skeleton. We show that one compound, PP121, blocks the proliferation of tumor cells by direct inhibition of oncogenic tyrosine kinases and phosphatidylinositol-3-OH kinases. These molecules demonstrate the feasibility of accessing a chemical space that intersects two families of oncogenes.
Figures
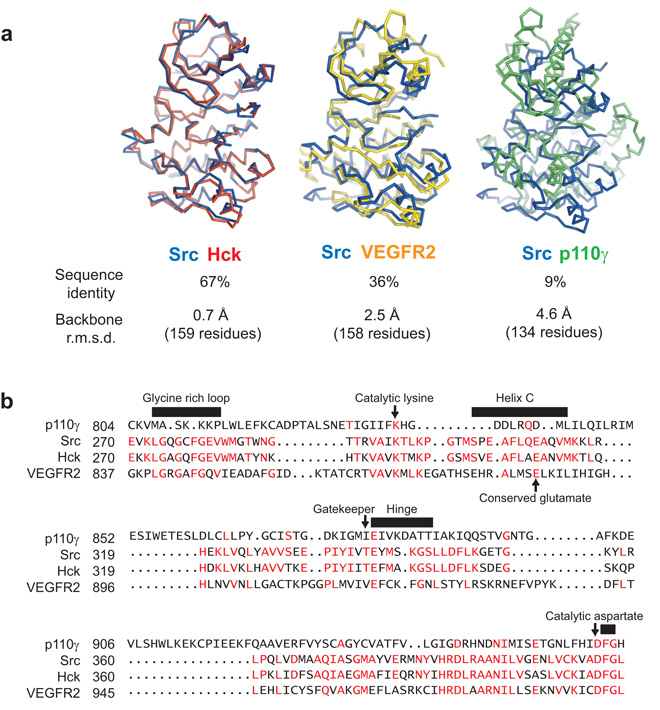
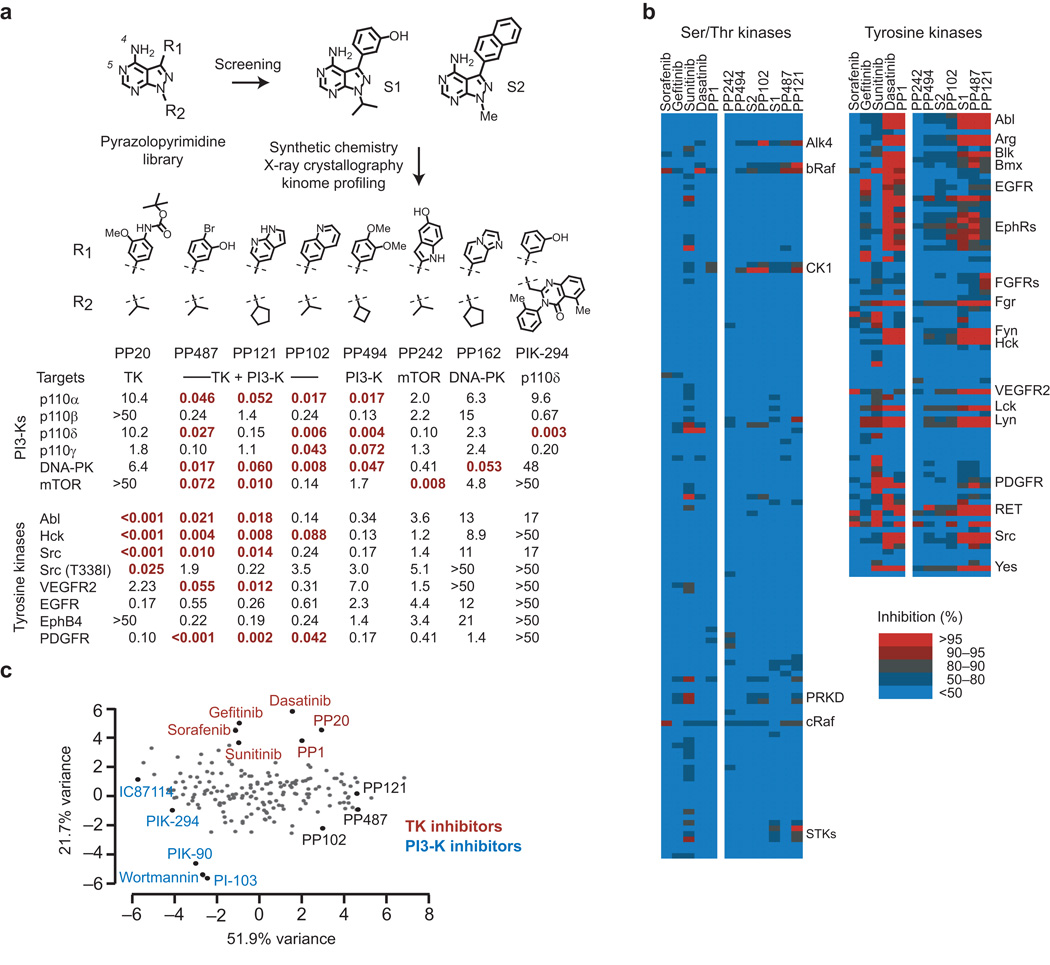
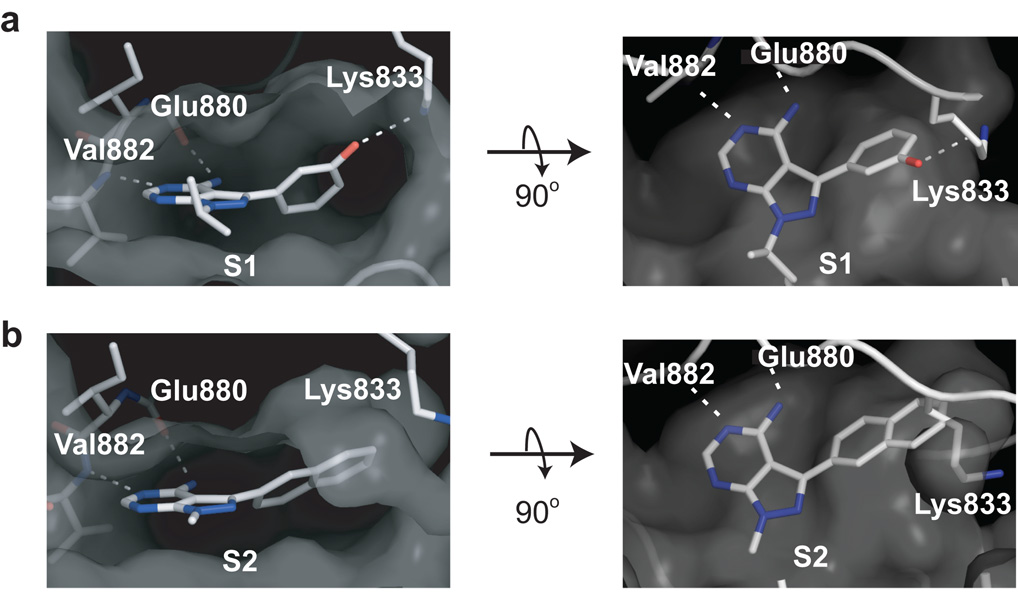
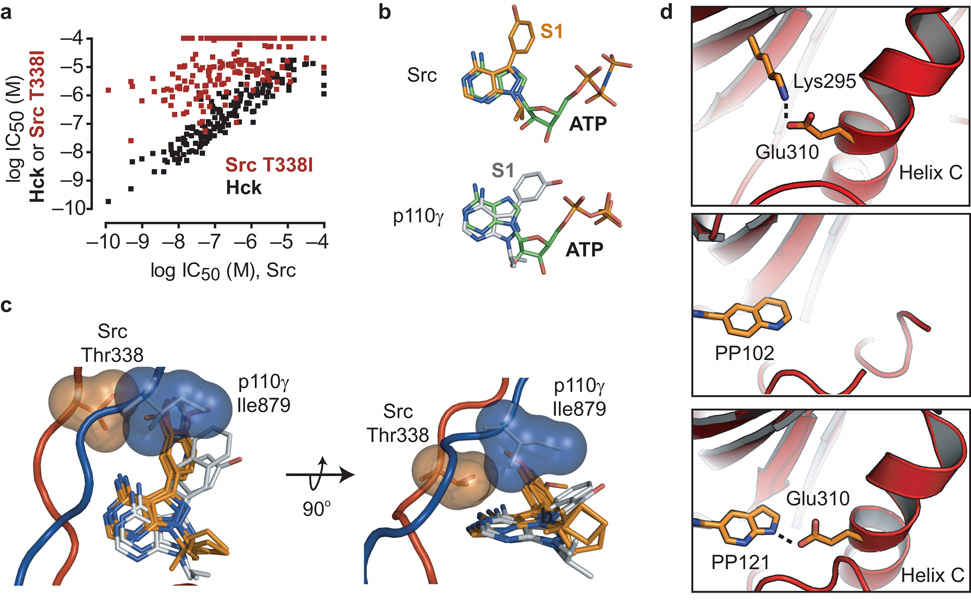
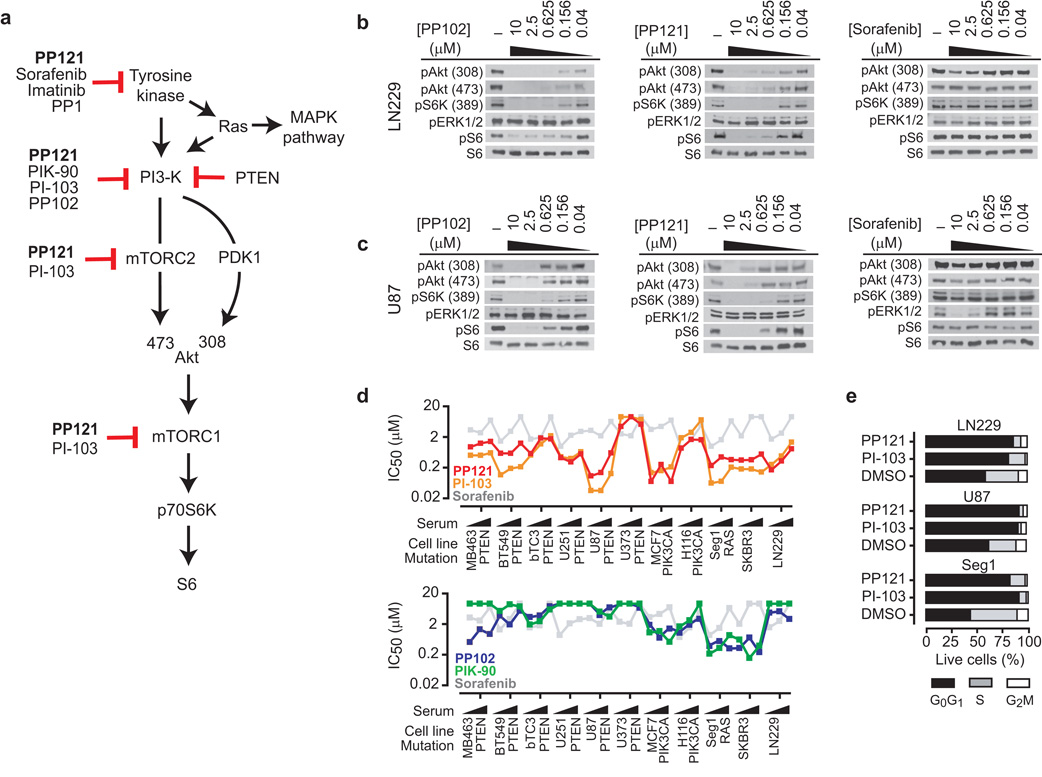
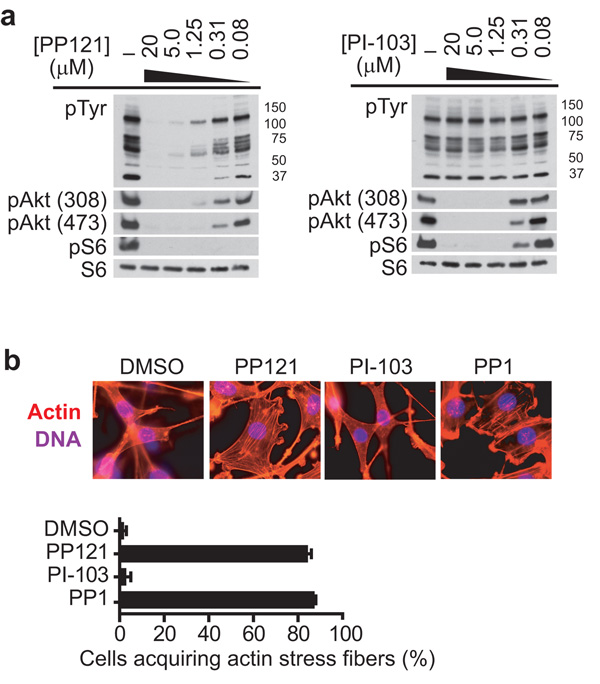
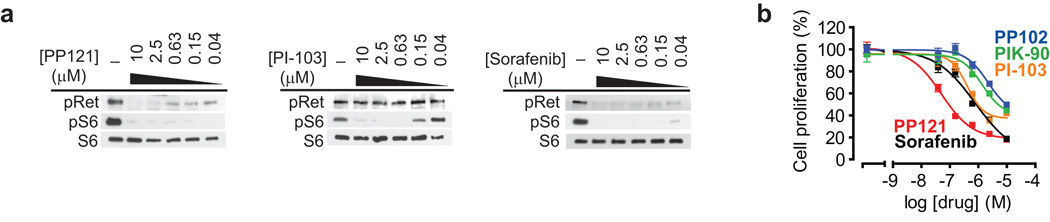
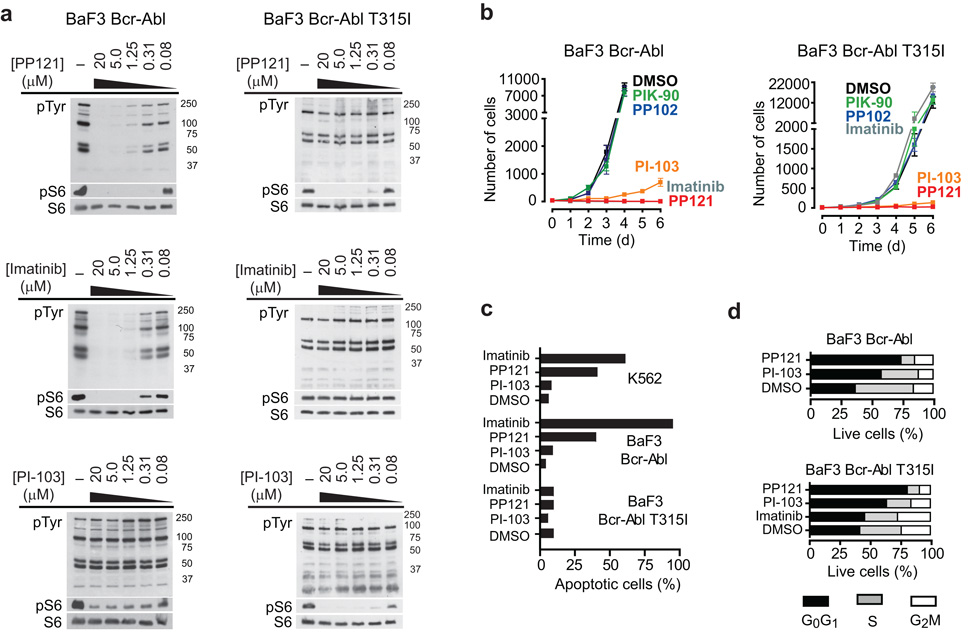
Comment in
-
Killing two kinase families with one stone.Nat Chem Biol. 2008 Nov;4(11):648-9. doi: 10.1038/nchembio1108-648. Nat Chem Biol. 2008. PMID: 18936744 No abstract available.
References
-
- Krause DS, Van Etten RA. Tyrosine kinases as targets for cancer therapy. N Engl J Med. 2005;353:172–187. - PubMed
-
- Sebolt-Leopold JS, English JM. Mechanisms of drug inhibition of signalling molecules. Nature. 2006;441:457–462. - PubMed
-
- Shaw RJ, Cantley LC. Ras, PI(3)K and mTOR signalling controls tumour cell growth. Nature. 2006;441:424–430. - PubMed
-
- Samuels Y, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. - PubMed
-
- Samuels Y, Velculescu VE. Oncogenic mutations of PIK3CA in human cancers. Cell Cycle. 2004;3:1221–1224. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
- Actions
- Actions
- Actions
- Actions
- PubChem-Substance/53799218
- PubChem-Substance/53799219
- PubChem-Substance/53799220
- PubChem-Substance/53799221
- PubChem-Substance/53799222
- PubChem-Substance/53799223
- PubChem-Substance/53799224
- PubChem-Substance/53799225
- PubChem-Substance/53799226
- PubChem-Substance/53799227
- PubChem-Substance/53799228
- PubChem-Substance/53799229
- PubChem-Substance/53799230
- PubChem-Substance/53799231
- PubChem-Substance/53799232
- PubChem-Substance/53799233
- PubChem-Substance/53799234
- PubChem-Substance/53799235
- PubChem-Substance/53799236
- PubChem-Substance/53799237
- PubChem-Substance/53799238
- PubChem-Substance/53799239
- PubChem-Substance/53799240
- PubChem-Substance/53799241
- PubChem-Substance/53799242
- PubChem-Substance/53799243
- PubChem-Substance/53799244
- PubChem-Substance/53799245
- PubChem-Substance/53799246
- PubChem-Substance/53799247
- PubChem-Substance/53799248
- PubChem-Substance/53799249
- PubChem-Substance/53799250
- PubChem-Substance/53799251
- PubChem-Substance/53799252
- PubChem-Substance/53799253
- PubChem-Substance/53799254
- PubChem-Substance/53799255
- PubChem-Substance/53799256
- PubChem-Substance/53799257
- PubChem-Substance/53799258
- PubChem-Substance/53799259
- PubChem-Substance/53799260
- PubChem-Substance/53799261
- PubChem-Substance/53799262
- PubChem-Substance/53799263
- PubChem-Substance/53799264
- PubChem-Substance/53799265
- PubChem-Substance/53799266
- PubChem-Substance/53799267
- PubChem-Substance/53799268
- PubChem-Substance/53799269
- PubChem-Substance/53799270
- PubChem-Substance/53799271
- PubChem-Substance/53799272
- PubChem-Substance/53799273
- PubChem-Substance/53799274
- PubChem-Substance/53799275
- PubChem-Substance/53799276
- PubChem-Substance/53799277
- PubChem-Substance/53799278
- PubChem-Substance/53799279
- PubChem-Substance/53799280
- PubChem-Substance/53799281
- PubChem-Substance/53799282
- PubChem-Substance/53799283
- PubChem-Substance/53799284
- PubChem-Substance/53799285
- PubChem-Substance/53799286
- PubChem-Substance/53799287
- PubChem-Substance/53799288
- PubChem-Substance/53799289
- PubChem-Substance/53799290
- PubChem-Substance/53799291
- PubChem-Substance/53799292
- PubChem-Substance/53799293
- PubChem-Substance/53799294
- PubChem-Substance/53799295
- PubChem-Substance/53799296
- PubChem-Substance/53799297
- PubChem-Substance/53799298
- PubChem-Substance/53799299
- PubChem-Substance/53799300
- PubChem-Substance/53799301
- PubChem-Substance/53799302
- PubChem-Substance/53799303
- PubChem-Substance/53799304
- PubChem-Substance/53799305
- PubChem-Substance/53799306
- PubChem-Substance/53799307
- PubChem-Substance/53799308
- PubChem-Substance/53799309
- PubChem-Substance/53799310
- PubChem-Substance/53799311
- PubChem-Substance/53799312
- PubChem-Substance/53799313
- PubChem-Substance/53799314
- PubChem-Substance/53799315
- PubChem-Substance/53799316
- PubChem-Substance/53799317
- PubChem-Substance/53799318
- PubChem-Substance/53799319
- PubChem-Substance/53799320
- PubChem-Substance/53799321
- PubChem-Substance/53799322
- PubChem-Substance/53799323
- PubChem-Substance/53799324
- PubChem-Substance/53799325
- PubChem-Substance/53799326
- PubChem-Substance/53799327
- PubChem-Substance/53799328
- PubChem-Substance/53799329
- PubChem-Substance/53799330
- PubChem-Substance/53799331
- PubChem-Substance/53799332
- PubChem-Substance/53799333
- PubChem-Substance/53799334
- PubChem-Substance/53799335
- PubChem-Substance/53799336
- PubChem-Substance/53799337
- PubChem-Substance/53799338
- PubChem-Substance/53799339
- PubChem-Substance/53799340
- PubChem-Substance/53799341
- PubChem-Substance/53799342
- PubChem-Substance/53799343
- PubChem-Substance/53799344
- PubChem-Substance/53799345
- PubChem-Substance/53799346
- PubChem-Substance/53799347
- PubChem-Substance/53799348
- PubChem-Substance/53799349
- PubChem-Substance/53799350
- PubChem-Substance/53799351
- PubChem-Substance/53799352
- PubChem-Substance/53799353
- PubChem-Substance/53799354
- PubChem-Substance/53799355
- PubChem-Substance/53799356
- PubChem-Substance/53799357
- PubChem-Substance/53799358
- PubChem-Substance/53799359
- PubChem-Substance/53799360
- PubChem-Substance/53799361
- PubChem-Substance/53799362
- PubChem-Substance/53799363
- PubChem-Substance/53799364
- PubChem-Substance/53799365
- PubChem-Substance/53799366
- PubChem-Substance/53799367
- PubChem-Substance/53799368
- PubChem-Substance/53799369
- PubChem-Substance/53799370
- PubChem-Substance/53799371
- PubChem-Substance/53799372
- PubChem-Substance/53799373
- PubChem-Substance/53799374
- PubChem-Substance/53799375
- PubChem-Substance/53799376
- PubChem-Substance/53799377
- PubChem-Substance/53799378
- PubChem-Substance/53799379
- PubChem-Substance/53799380
- PubChem-Substance/53799381
- PubChem-Substance/53799382
- PubChem-Substance/53799383
- PubChem-Substance/53799384
- PubChem-Substance/53799385
- PubChem-Substance/53799386
- PubChem-Substance/53799387
- PubChem-Substance/53799388
- PubChem-Substance/53799389
- PubChem-Substance/53799390
- PubChem-Substance/53799391
- PubChem-Substance/53799392
- PubChem-Substance/53799393
- PubChem-Substance/53799394
- PubChem-Substance/53799395
- PubChem-Substance/53799396
- PubChem-Substance/53799397
- PubChem-Substance/53799398
- PubChem-Substance/53799399
- PubChem-Substance/53799400
- PubChem-Substance/53799401
- PubChem-Substance/53799402
- PubChem-Substance/53799403
- PubChem-Substance/53799404
- PubChem-Substance/53799405
- PubChem-Substance/53799406
- PubChem-Substance/53799407
- PubChem-Substance/53799408
- PubChem-Substance/53799409
- PubChem-Substance/53799410
- PubChem-Substance/53799411
- PubChem-Substance/53799412
- PubChem-Substance/53799413
- PubChem-Substance/53799414
- PubChem-Substance/53799415
- PubChem-Substance/53799416
- PubChem-Substance/53799417
- PubChem-Substance/53799418
- PubChem-Substance/53799419
- PubChem-Substance/53799420
- PubChem-Substance/53799421
- PubChem-Substance/53799422
- PubChem-Substance/53799423
- PubChem-Substance/53799424
- PubChem-Substance/53799425
- PubChem-Substance/53799426
- PubChem-Substance/53799427
- PubChem-Substance/53799428
- PubChem-Substance/53799429
- PubChem-Substance/53799430
- PubChem-Substance/53799431
- PubChem-Substance/53799432
- PubChem-Substance/53799433
- PubChem-Substance/53799434
- PubChem-Substance/53799435
- PubChem-Substance/53799436
- PubChem-Substance/53799437
- PubChem-Substance/53799438
- PubChem-Substance/53799439
- PubChem-Substance/53799440
- PubChem-Substance/53799441
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Chemical Information
Molecular Biology Databases
Miscellaneous
