

Modelling heterotachy in phylogenetic inference by reversible-jump Markov chain Monte Carlo
- PMID: 18852097
- PMCID: PMC2607421
- DOI: 10.1098/rstb.2008.0178
Modelling heterotachy in phylogenetic inference by reversible-jump Markov chain Monte Carlo
Abstract
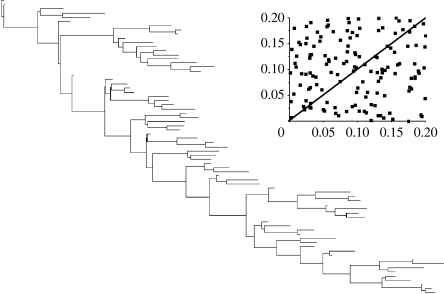
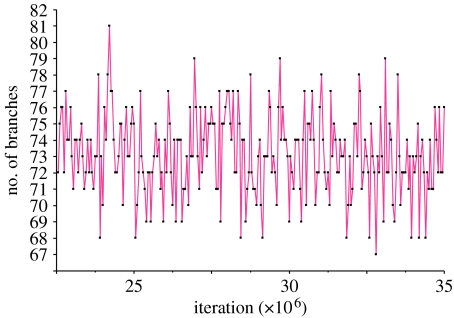
The rate at which a given site in a gene sequence alignment evolves over time may vary. This phenomenon--known as heterotachy--can bias or distort phylogenetic trees inferred from models of sequence evolution that assume rates of evolution are constant. Here, we describe a phylogenetic mixture model designed to accommodate heterotachy. The method sums the likelihood of the data at each site over more than one set of branch lengths on the same tree topology. A branch-length set that is best for one site may differ from the branch-length set that is best for some other site, thereby allowing different sites to have different rates of change throughout the tree. Because rate variation may not be present in all branches, we use a reversible-jump Markov chain Monte Carlo algorithm to identify those branches in which reliable amounts of heterotachy occur. We implement the method in combination with our 'pattern-heterogeneity' mixture model, applying it to simulated data and five published datasets. We find that complex evolutionary signals of heterotachy are routinely present over and above variation in the rate or pattern of evolution across sites, that the reversible-jump method requires far fewer parameters than conventional mixture models to describe it, and serves to identify the regions of the tree in which heterotachy is most pronounced. The reversible-jump procedure also removes the need for a posteriori tests of 'significance' such as the Akaike or Bayesian information criterion tests, or Bayes factors. Heterotachy has important consequences for the correct reconstruction of phylogenies as well as for tests of hypotheses that rely on accurate branch-length information. These include molecular clocks, analyses of tempo and mode of evolution, comparative studies and ancestral state reconstruction. The model is available from the authors' website, and can be used for the analysis of both nucleotide and morphological data.
Figures



References
-
- Akaike H. A new look at the statistical model identification. Automatic Control, IEEE Transactions on. 1974;19:716–723. doi:10.1109/TAC.1974.1100705 - DOI
-
- Ane C, Burleigh J.G, McMahon M.M, Sanderson M.J. Covarion structure in plastid genome evolution: a new statistical test. Mol. Biol. Evol. 2005;22:914–924. doi:10.1093/molbev/msi076 - DOI - PubMed
-
- Blackburne, B. P., Hay, A. J. & Goldstein, R. A. 2008 Changing selective pressures during antigenic changes in Human influenza H3. PLoS Pathog 4, e1000058. (doi:10.1371/journal.ppat.1000058) - DOI - PMC - PubMed
-
- Boys R.J, Henderson D.A. A comparison of reversible jump MCMC algorithms for DNA sequence segmentation using hidden Markov models. Comput. Sci. Stat. 2001;33:35–49.
-
- Buchheim M.A, Michalopulos E.A, Buchheim J.A. Phylogeny of the Chlorophyceae with special reference to the Sphaeropleales: a study of 18S and 26S rDNA data. J. Phycol. 2001;37:819–835. doi:10.1046/j.1529-8817.2001.00162.x - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources