

Basing population genetic inferences and models of molecular evolution upon desired stationary distributions of DNA or protein sequences
- PMID: 18852105
- PMCID: PMC2607412
- DOI: 10.1098/rstb.2008.0167
Basing population genetic inferences and models of molecular evolution upon desired stationary distributions of DNA or protein sequences
Abstract
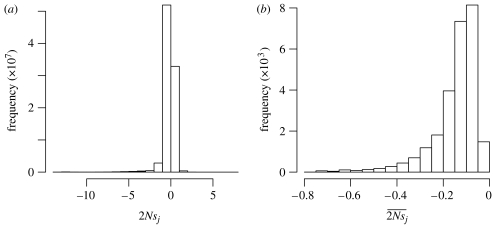
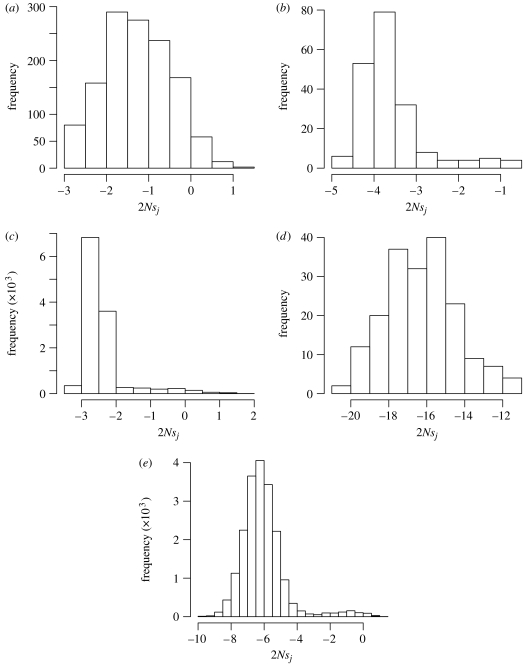
Models of molecular evolution tend to be overly simplistic caricatures of biology that are prone to assigning high probabilities to biologically implausible DNA or protein sequences. Here, we explore how to construct time-reversible evolutionary models that yield stationary distributions of sequences that match given target distributions. By adopting comparatively realistic target distributions,evolutionary models can be improved. Instead of focusing on estimating parameters, we concentrate on the population genetic implications of these models. Specifically, we obtain estimates of the product of effective population size and relative fitness difference of alleles. The approach is illustrated with two applications to protein-coding DNA. In the first, a codon-based evolutionary model yields a stationary distribution of sequences, which, when the sequences are translated,matches a variable-length Markov model trained on human proteins. In the second, we introduce an insertion-deletion model that describes selectively neutral evolutionary changes to DNA. We then show how to modify the neutral model so that its stationary distribution at the amino acid level can match a profile hidden Markov model, such as the one associated with the Pfam database.
Figures
Similar articles
-
General continuous-time Markov model of sequence evolution via insertions/deletions: are alignment probabilities factorable?BMC Bioinformatics. 2016 Aug 11;17:304. doi: 10.1186/s12859-016-1105-7. BMC Bioinformatics. 2016. PMID: 27638547 Free PMC article.
-
GENOMEPOP: a program to simulate genomes in populations.BMC Bioinformatics. 2008 Apr 30;9:223. doi: 10.1186/1471-2105-9-223. BMC Bioinformatics. 2008. PMID: 18447924 Free PMC article.
-
The nearly neutral and selection theories of molecular evolution under the fisher geometrical framework: substitution rate, population size, and complexity.Genetics. 2012 Jun;191(2):523-34. doi: 10.1534/genetics.112.138628. Epub 2012 Mar 16. Genetics. 2012. PMID: 22426879 Free PMC article.
-
The modern molecular clock.Nat Rev Genet. 2003 Mar;4(3):216-24. doi: 10.1038/nrg1020. Nat Rev Genet. 2003. PMID: 12610526 Review.
-
Protein evolution depends on multiple distinct population size parameters.BMC Evol Biol. 2018 Feb 8;18(1):17. doi: 10.1186/s12862-017-1085-x. BMC Evol Biol. 2018. PMID: 29422024 Free PMC article. Review.
Cited by
-
All-at-once RNA folding with 3D motif prediction framed by evolutionary information.bioRxiv [Preprint]. 2025 Apr 8:2024.12.17.628809. doi: 10.1101/2024.12.17.628809. bioRxiv. 2025. PMID: 39764046 Free PMC article. Preprint.
-
All-at-once RNA folding with 3D motif prediction framed by evolutionary information.Res Sq [Preprint]. 2025 Mar 26:rs.3.rs-5664139. doi: 10.21203/rs.3.rs-5664139/v1. Res Sq. 2025. PMID: 40195991 Free PMC article. Preprint.
-
Fast optimization of statistical potentials for structurally constrained phylogenetic models.BMC Evol Biol. 2009 Sep 9;9:227. doi: 10.1186/1471-2148-9-227. BMC Evol Biol. 2009. PMID: 19740424 Free PMC article.
-
Mutation-selection models of coding sequence evolution with site-heterogeneous amino acid fitness profiles.Proc Natl Acad Sci U S A. 2010 Mar 9;107(10):4629-34. doi: 10.1073/pnas.0910915107. Epub 2010 Feb 22. Proc Natl Acad Sci U S A. 2010. PMID: 20176949 Free PMC article.
-
A formal perturbation equation between genotype and phenotype determines the Evolutionary Action of protein-coding variations on fitness.Genome Res. 2014 Dec;24(12):2050-8. doi: 10.1101/gr.176214.114. Epub 2014 Sep 12. Genome Res. 2014. PMID: 25217195 Free PMC article.
References
-
- Bejerano G. Algorithms for variable length Markov chain modeling. Bioinformatics. 2004;20:788–789. doi:10.1093/bioinformatics/btg489 - DOI - PubMed
-
- Bejerano G, Yona G. Variations on probabilistic suffix trees: statistical modeling and prediction of protein families. Bioinformatics. 2001;17:23–43. doi:10.1093/bioinformatics/17.1.23 - DOI - PubMed
-
- Berg J, Willmann S, Lässig M. Adaptive evolution of transcription factor binding sites. BMC Evol. Biol. 2004;4:42. doi:10.1186/1471-2148-4-42 - DOI - PMC - PubMed
-
- Blanquart S, Lartillot N. A Bayesian compound stochastic process for modeling nonstationary and nonhomogeneous sequence evolution. Mol. Biol. Evol. 2006;23:2058–2071. doi:10.1093/molbev/msl091 - DOI - PubMed
-
- Blanquart S, Lartillot N. A site- and time-heterogeneous model of amino acid replacement. Mol. Biol. Evol. 2008;25:842–858. doi:10.1093/molbev/msn018 - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
