

Accounting for ancestry: population substructure and genome-wide association studies
- PMID: 18852203
- PMCID: PMC2782357
- DOI: 10.1093/hmg/ddn268
Accounting for ancestry: population substructure and genome-wide association studies
Abstract
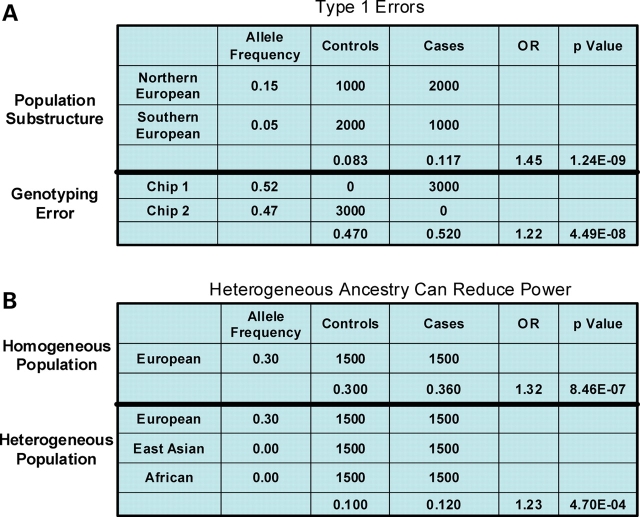
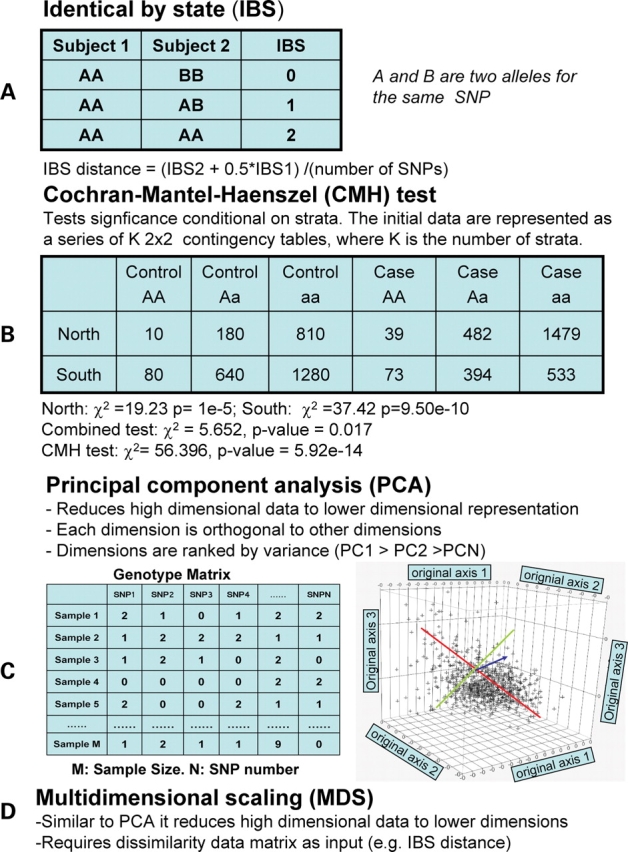
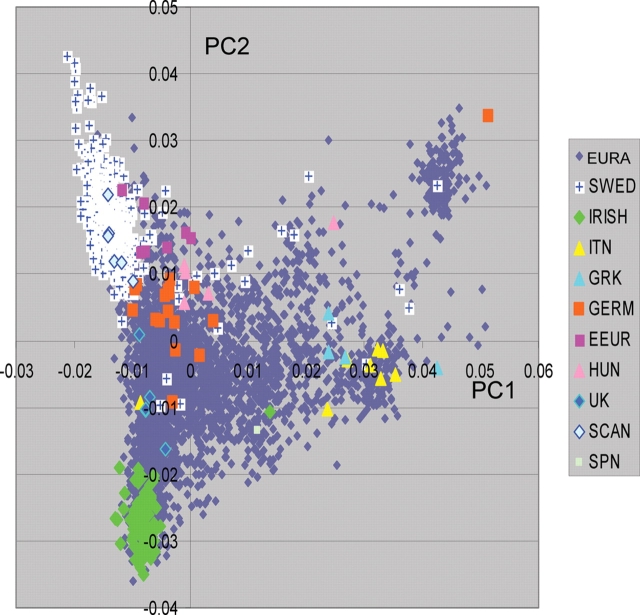
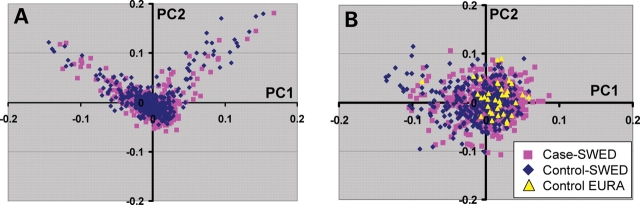
Accounting for the genetic substructure of human populations has become a major practical issue for studying complex genetic disorders. Allele frequency differences among ethnic groups and subgroups and admixture between different ethnic groups can result in frequent false-positive results or reduced power in genetic studies. Here, we review the problems and progress in defining population differences and the application of statistical methods to improve association studies. It is now possible to take into account the confounding effects of population stratification using thousands of unselected genome-wide single-nucleotide polymorphisms or, alternatively, selected panels of ancestry informative markers. These methods do not require any demographic information and therefore can be widely applied to genotypes available from multiple sources. We further suggest that it will be important to explore results in homogeneous population subsets as we seek to define the extent to which genomic variation influences complex phenotypes.
Figures
References
-
- Hom G., Graham R.R., Modrek B., Taylor K.E., Ortmann W., Garnier S., Lee A.T., Chung S.A., Ferreira R.C., Pant P.V., et al. Association of systemic lupus erythematosus with C8orf13-BLK and ITGAM-ITGAX. N. Engl. J. Med. 2008;358:900–909. - PubMed
-
- Laird N.M., Lange C. Family-based designs in the age of large-scale gene-association studies. Nat. Rev. Genet. 2006;7:385–394. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
