

The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage
- PMID: 18852295
- PMCID: PMC2568022
- DOI: 10.1083/jcb.200711150
The HINT1 tumor suppressor regulates both gamma-H2AX and ATM in response to DNA damage
Abstract
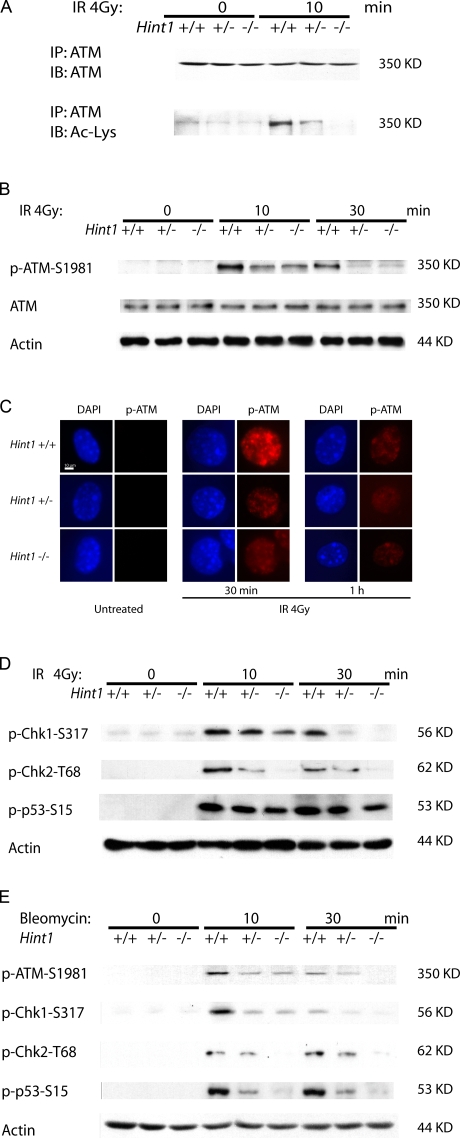
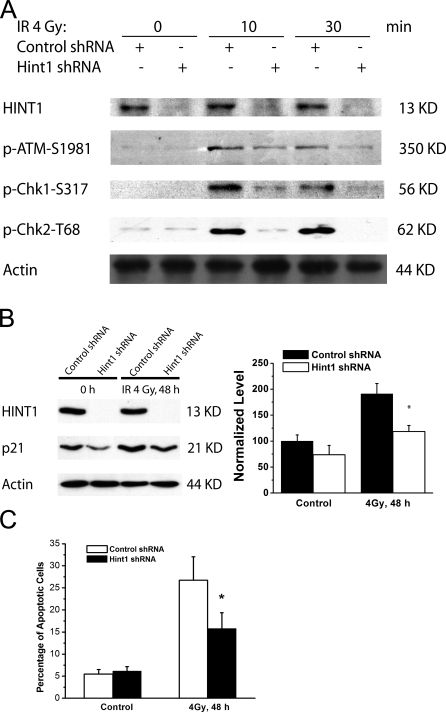
Hint1 is a haploinsufficient tumor suppressor gene and the underlying molecular mechanisms for its tumor suppressor function are unknown. In this study we demonstrate that HINT1 participates in ionizing radiation (IR)-induced DNA damage responses. In response to IR, HINT1 is recruited to IR-induced foci (IRIF) and associates with gamma-H2AX and ATM. HINT1 deficiency does not affect the formation of gamma-H2AX foci; however, it impairs the removal of gamma-H2AX foci after DNA damage and this is associated with impaired acetylation of gamma-H2AX. HINT1 deficiency also impairs acetylation of ATM and activation of ATM and its downstream effectors, and retards DNA repair, in response to IR. HINT1-deficient cells exhibit resistance to IR-induced apoptosis and several types of chromosomal abnormalities. Our findings suggest that the tumor suppressor function of HINT1 is caused by, at least in part, its normal role in enhancing cellular responses to DNA damage by regulating the functions of both gamma-H2AX and ATM.
Figures





References
-
- Ahel, I., U. Rass, S.F. El-Khamisy, S. Katyal, P.M. Clements, P.J. McKinnon, K.W. Caldecott, and S.C. West. 2006. The neurodegenerative disease protein aprataxin resolves abortive DNA ligation intermediates. Nature. 443:713–716. - PubMed
-
- Ajit, S.K., S. Ramineni, W. Edris, R.A. Hunt, W.T. Hum, J.R. Hepler, and K.H. Young. 2007. RGSZ1 interacts with protein kinase C interacting protein PKCI-1 and modulates mu opioid receptor signaling. Cell. Signal. 19:723–730. - PubMed
-
- Bakkenist, C.J., and M.B. Kastan. 2003. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 421:499–506. - PubMed
-
- Balajee, A.S., and C.R. Geard. 2004. Replication protein A and gamma-H2AX foci assembly is triggered by cellular response to DNA double-strand breaks. Exp. Cell Res. 300:320–334. - PubMed
-
- Barlow, C., K.D. Brown, C.X. Deng, D.A. Tagle, and A. Wynshaw-Boris. 1997. Atm selectively regulates distinct p53-dependent cell-cycle checkpoint and apoptotic pathways. Nat. Genet. 17:453–456. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
