

Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice
- PMID: 18852886
- PMCID: PMC2559862
- DOI: 10.1371/journal.pone.0003387
Functional deficits in nNOSmu-deficient skeletal muscle: myopathy in nNOS knockout mice
Abstract
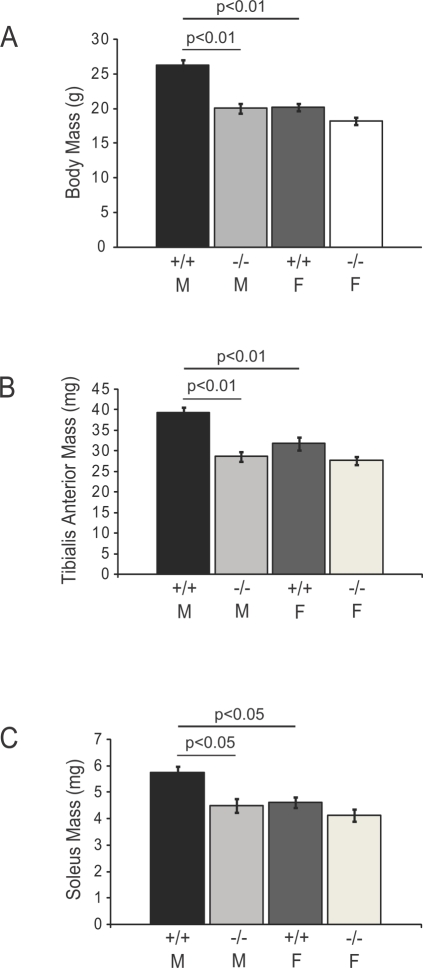
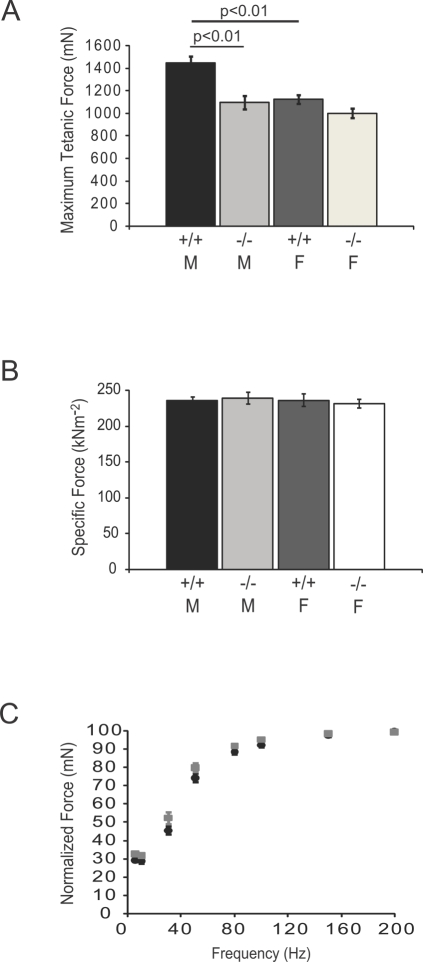
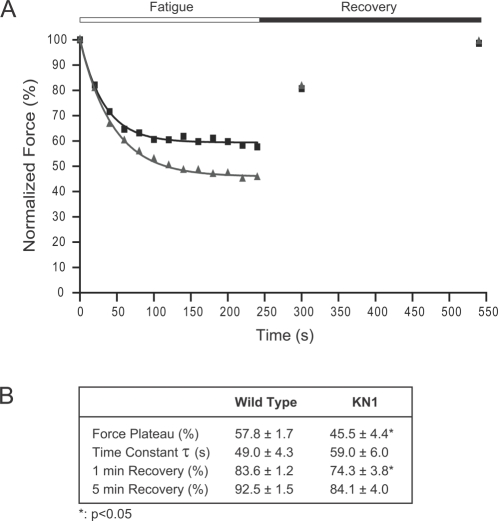
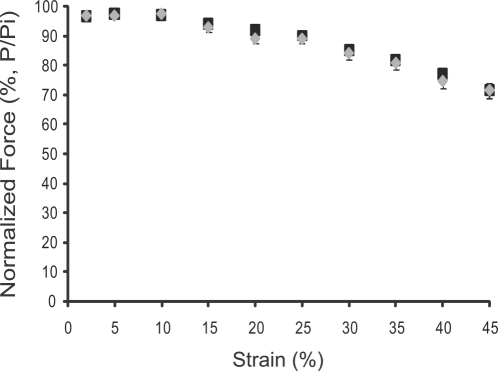
Skeletal muscle nNOSmu (neuronal nitric oxide synthase mu) localizes to the sarcolemma through interaction with the dystrophin-associated glycoprotein (DAG) complex, where it synthesizes nitric oxide (NO). Disruption of the DAG complex occurs in dystrophinopathies and sarcoglycanopathies, two genetically distinct classes of muscular dystrophy characterized by progressive loss of muscle mass, muscle weakness and increased fatigability. DAG complex instability leads to mislocalization and downregulation of nNOSmu; but this is thought to play a minor role in disease pathogenesis. This view persists without knowledge of the role of nNOS in skeletal muscle contractile function in vivo and has influenced gene therapy approaches to dystrophinopathy, the majority of which do not restore sarcolemmal nNOSmu. We address this knowledge gap by evaluating skeletal muscle function in nNOS knockout (KN1) mice using an in situ approach, in which the muscle is maintained in its normal physiological environment. nNOS-deficiency caused reductions in skeletal muscle bulk and maximum tetanic force production in male mice only. Furthermore, nNOS-deficient muscles from both male and female mice exhibited increased susceptibility to contraction-induced fatigue. These data suggest that aberrant nNOSmu signaling can negatively impact three important clinical features of dystrophinopathies and sarcoglycanopathies: maintenance of muscle bulk, force generation and fatigability. Our study suggests that restoration of sarcolemmal nNOSmu expression in dystrophic muscles may be more important than previously appreciated and that it should be a feature of any fully effective gene therapy-based intervention.
Conflict of interest statement
Figures
References
-
- Stamler JS, Meissner G. Physiology of nitric oxide in skeletal muscle. Physiol Rev. 2001;81:209–237. - PubMed
-
- Silvagno F, Xia H, Bredt DS. Neuronal nitric-oxide synthase-mu, an alternatively spliced isoform expressed in differentiated skeletal muscle. J Biol Chem. 1996;271:11204–11208. - PubMed
-
- Kobzik L, Reid MB, Bredt DS, Stamler JS. Nitric oxide in skeletal muscle. Nature. 1994;372:546–548. - PubMed
-
- Reid MB, Kobzik L, Bredt DS, Stamler JS. Nitric oxide modulates excitation-contraction coupling in the diaphragm. Comp Biochem Physiol A Mol Integr Physiol. 1998;19:211–218. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
