

Selected reaction monitoring for quantitative proteomics: a tutorial
- PMID: 18854821
- PMCID: PMC2583086
- DOI: 10.1038/msb.2008.61
Selected reaction monitoring for quantitative proteomics: a tutorial
Abstract
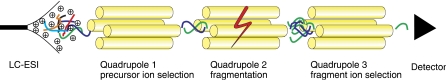
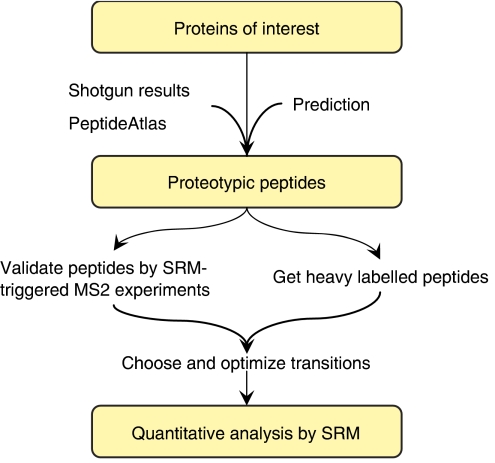
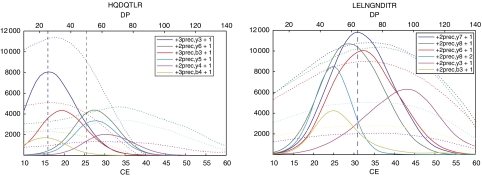
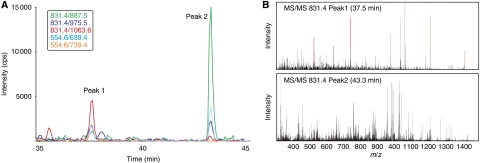
Systems biology relies on data sets in which the same group of proteins is consistently identified and precisely quantified across multiple samples, a requirement that is only partially achieved by current proteomics approaches. Selected reaction monitoring (SRM)-also called multiple reaction monitoring-is emerging as a technology that ideally complements the discovery capabilities of shotgun strategies by its unique potential for reliable quantification of analytes of low abundance in complex mixtures. In an SRM experiment, a predefined precursor ion and one of its fragments are selected by the two mass filters of a triple quadrupole instrument and monitored over time for precise quantification. A series of transitions (precursor/fragment ion pairs) in combination with the retention time of the targeted peptide can constitute a definitive assay. Typically, a large number of peptides are quantified during a single LC-MS experiment. This tutorial explains the application of SRM for quantitative proteomics, including the selection of proteotypic peptides and the optimization and validation of transitions. Furthermore, normalization and various factors affecting sensitivity and accuracy are discussed.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
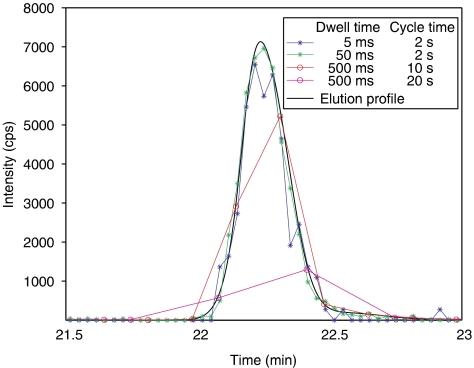
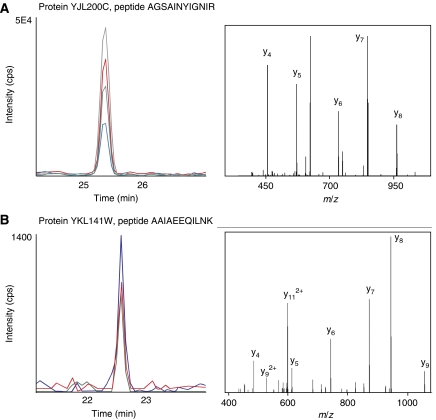
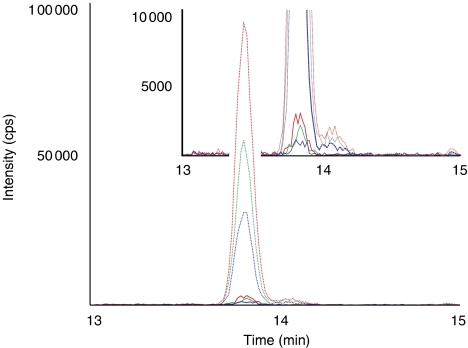
References
-
- Ashburner M, Ball CA, Blake JA, Botstein D, Butler H, Cherry JM, Davis AP, Dolinski K, Dwight SS, Eppig JT, Harris MA, Hill DP, Issel-Tarver L, Kasarskis A, Lewis S, Matese JC, Richardson JE, Ringwald M, Rubin GM, Sherlock G (2000) Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25: 25–29 - PMC - PubMed
-
- Barr JR, Maggio VL, Patterson DG Jr, Cooper GR, Henderson LO, Turner WE, Smith SJ, Hannon WH, Needham LL, Sampson EJ (1996) Isotope dilution–mass spectrometric quantification of specific proteins: model application with apolipoprotein A-I. Clin Chem 42: 1676–1682 - PubMed
-
- Beynon RJ, Doherty MK, Pratt JM, Gaskell SJ (2005) Multiplexed absolute quantification in proteomics using artificial QCAT proteins of concatenated signature peptides. Nat Methods 2: 587–589 - PubMed
-
- Bodenmiller B, Malmstrom J, Gerrits B, Campbell D, Lam H, Schmidt A, Rinner O, Mueller LN, Shannon PT, Pedrioli PG, Panse C, Lee HK, Schlapbach R, Aebersold R (2007) PhosphoPep–a phosphoproteome resource for systems biology research in Drosophila Kc167 cells. Mol Syst Biol 3: 139 10.1038/msb4100182 - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
