

Congenital hereditary endothelial dystrophy with progressive sensorineural deafness (Harboyan syndrome)
- PMID: 18922146
- PMCID: PMC2576053
- DOI: 10.1186/1750-1172-3-28
Congenital hereditary endothelial dystrophy with progressive sensorineural deafness (Harboyan syndrome)
Abstract
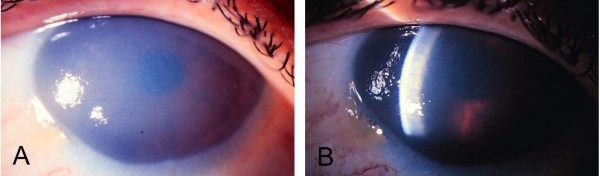
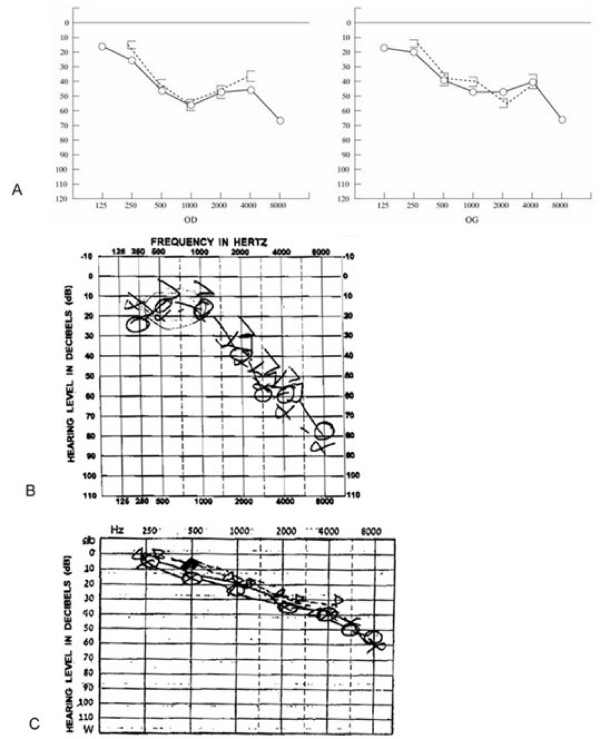
Harboyan syndrome is a degenerative corneal disorder defined as congenital hereditary endothelial dystrophy (CHED) accompanied by progressive, postlingual sensorineural hearing loss. To date, 24 cases from 11 families of various origin (Asian Indian, South American Indian, Sephardi Jewish, Brazilian Portuguese, Dutch, Gypsy, Moroccan, Dominican) have been reported. More than 50% of the reported cases have been associated with parental consanguinity. The ocular manifestations in Harboyan syndrome include diffuse bilateral corneal edema occurring with severe corneal clouding, blurred vision, visual loss and nystagmus. They are apparent at birth or within the neonatal period and are indistinguishable from those characteristic of the autosomal recessive CHED (CHED2). Hearing deficit in Harboyan is slowly progressive and typically found in patients 10-25 years old. There are no reported cases with prelinglual deafness, however, a significant hearing loss in children as young as 4 years old has been detected by audiometry, suggesting that hearing may be affected earlier, even at birth. Harboyan syndrome is caused by mutations in the SLC4A11 gene located at the CHED2 locus on chromosome 20p13-p12, indicating that CHED2 and Harboyan syndrome are allelic disorders. A total of 62 different SLC4A11 mutations have been reported in 98 families (92 CHED2 and 6 Harboyan). All reported cases have been consistent with autosomal recessive transmission. Diagnosis is based on clinical criteria, detailed ophthalmological assessment and audiometry. A molecular confirmation of the clinical diagnosis is feasible. A variety of genetic, metabolic, developmental and acquired diseases presenting with clouding of the cornea should be considered in the differential diagnosis (Peters anomaly, sclerocornea, limbal dermoids, congenital glaucoma). Audiometry must be performed to differentiate Harboyan syndrome from CHED2. Autosomal recessive types of CHED (CHED2 and Harboyan syndrome) should carefully be distinguished from the less severe autosomal dominant type CHED1. The ocular abnormalities in patients with Harboyan syndrome may be treated with topical hyperosmolar solutions. However, corneal transplantation (penetrating keratoplasty) represents definitive treatment. Corneal transplantation produces a substantial visual gain and has a relatively good surgical prognosis. Audiometric monitoring should be offered to all patients with CHED2. Hearing aids may be necessary in adolescence.
Figures
Similar articles
-
Absence of phenotype-genotype correlation of patients expressing mutations in the SLC4A11 gene.Cornea. 2010 Mar;29(3):302-6. doi: 10.1097/ICO.0b013e3181ae9038. Cornea. 2010. PMID: 20118786
-
Borate transporter SLC4A11 mutations cause both Harboyan syndrome and non-syndromic corneal endothelial dystrophy.J Med Genet. 2007 May;44(5):322-6. doi: 10.1136/jmg.2006.046904. Epub 2007 Jan 12. J Med Genet. 2007. PMID: 17220209 Free PMC article.
-
Mutational spectrum of the SLC4A11 gene in autosomal recessive congenital hereditary endothelial dystrophy.Mol Vis. 2007 Jul 26;13:1327-32. Mol Vis. 2007. PMID: 17679935
-
SLC4A11 and the Pathophysiology of Congenital Hereditary Endothelial Dystrophy.Biomed Res Int. 2015;2015:475392. doi: 10.1155/2015/475392. Epub 2015 Sep 16. Biomed Res Int. 2015. PMID: 26451371 Free PMC article. Review.
-
[The revised newest IC³D classification of corneal dystrophies].Klin Monbl Augenheilkd. 2015 Mar;232(3):283-94. doi: 10.1055/s-0041-100774. Epub 2015 Mar 24. Klin Monbl Augenheilkd. 2015. PMID: 25803558 Review. German.
Cited by
-
Classification of posterior polymorphous corneal dystrophy as a corneal ectatic disorder following confirmation of associated significant corneal steepening.JAMA Ophthalmol. 2013 Dec;131(12):1583-90. doi: 10.1001/jamaophthalmol.2013.5036. JAMA Ophthalmol. 2013. PMID: 24113819 Free PMC article.
-
Corneal dystrophies.Nat Rev Dis Primers. 2020 Jun 11;6(1):46. doi: 10.1038/s41572-020-0178-9. Nat Rev Dis Primers. 2020. PMID: 32528047 Review.
-
[Corneal disease in childhood-Hereditary, degenerative or infectious?].Ophthalmologie. 2023 Aug;120(8):811-817. doi: 10.1007/s00347-023-01897-3. Epub 2023 Jul 12. Ophthalmologie. 2023. PMID: 37438454 Review. German.
-
SLC4A11 depletion impairs NRF2 mediated antioxidant signaling and increases reactive oxygen species in human corneal endothelial cells during oxidative stress.Sci Rep. 2017 Jun 22;7(1):4074. doi: 10.1038/s41598-017-03654-4. Sci Rep. 2017. PMID: 28642546 Free PMC article.
-
Ptosis, ophthalmoplegia and corneal endothelial disease - ocular manifestations of mitochondrial disease.Am J Ophthalmol Case Rep. 2021 Mar 17;22:101073. doi: 10.1016/j.ajoc.2021.101073. eCollection 2021 Jun. Am J Ophthalmol Case Rep. 2021. PMID: 33869891 Free PMC article.
References
-
- Waring GO, 3rd, Bourne WM, Edelhauser HF, Kenyon KR. The corneal endothelium. Normal and pathologic structure and function. Ophthalmology. 1982;89:531–590. - PubMed
-
- Vithana EN, Morgan P, Sundaresan P, Ebenezer ND, Tan DT, Mohamed MD, Anand S, Khine KO, Venkataraman D, Yong VH, Salto-Tellez M, Venkatraman A, Guo K, Hemadevi B, Srinivasan M, Prajna V, Khine M, Casey JR, Inglehearn CF, Aung T. Mutations in sodium-borate cotransporter SLC4A11 cause recessive congenital hereditary endothelial dystrophy (CHED2) Nat Genet. 2006;38:755–757. doi: 10.1038/ng1824. - DOI - PubMed
-
- Héon E, Greenberg A, Kopp KK, Rootman D, Vincent AL, Billingsley G, Priston M, Dorval KM, Chow RL, McInnes RR, Heathcote G, Westall C, Sutphin JE, Semina E, Bremner R, Stone EM. VSX1: a gene for posterior polymorphous dystrophy and keratoconus. Hum Mol Genet. 2002;11:1029–1036. doi: 10.1093/hmg/11.9.1029. - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
