

Dual inhibition of PI3Kalpha and mTOR as an alternative treatment for Kaposi's sarcoma
- PMID: 18922908
- PMCID: PMC2645787
- DOI: 10.1158/0008-5472.CAN-08-0878
Dual inhibition of PI3Kalpha and mTOR as an alternative treatment for Kaposi's sarcoma
Abstract
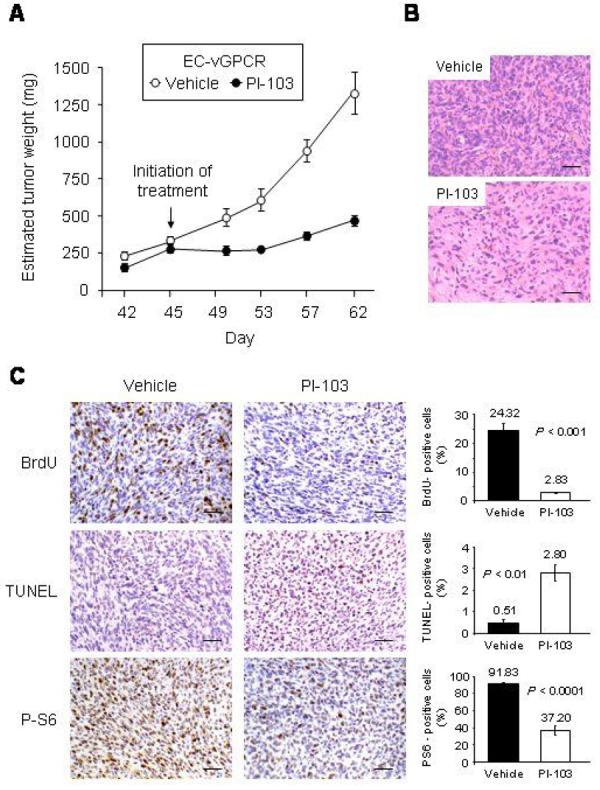
Rapamycin (or sirolimus), the prototypical inhibitor of the mammalian target of rapamycin (mTOR) and an immunosuppressant used for the prevention of renal transplant rejection, has recently emerged as an effective treatment for Kaposi's sarcoma (KS), an enigmatic vascular tumor and a model for pathologic angiogenesis. Indeed, recent work supports a role for mTOR as a central player in the transformation of endothelial cells by the KS-associated herpesvirus-encoded G protein-coupled receptor (vGPCR), the viral oncogene believed to be responsible for causing KS. However, emerging evidence that rapamycin may transiently promote the activation of Akt may limit its use as an anti-KS therapy. Here, we show that activation of Akt in endothelial cells expressing vGPCR is augmented by treatment with rapamycin, resulting in the up-regulation of several Akt proliferative and survival pathways. However, use of a novel dual phosphatidylinositol 3-kinase alpha (PI3Kalpha)/mTOR inhibitor, PI-103, effectively and independently blocked activation of both PI3K and mTOR in vGPCR-expressing endothelial cells. This resulted in more effective inhibition of endothelial cell proliferation and survival in vitro and tumor growth in vivo. Our results suggest that PI-103 may be an effective therapeutic option for the treatment of patients with KS. Moreover, as KS may serve as a model for pathologic angiogenesis, our results further provide the basis for the early assessment of PI-103 as an antiangiogenic chemotherapeutic.
Figures
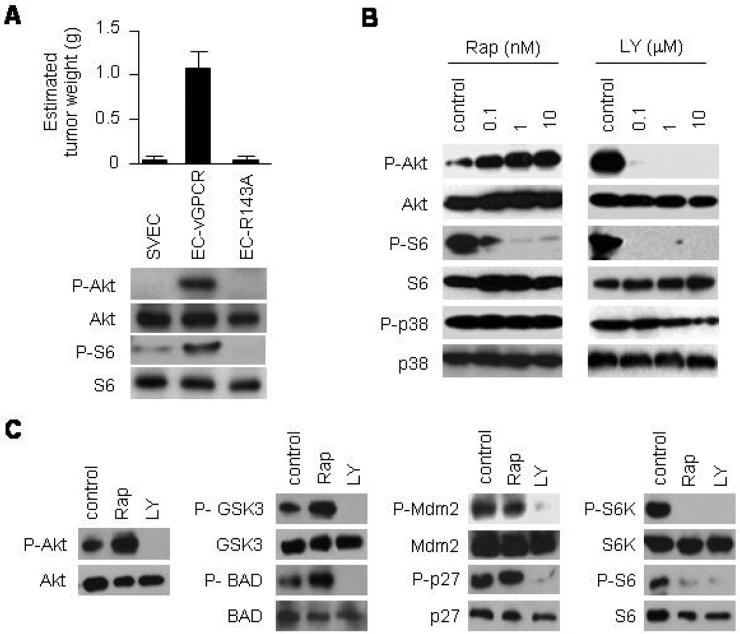
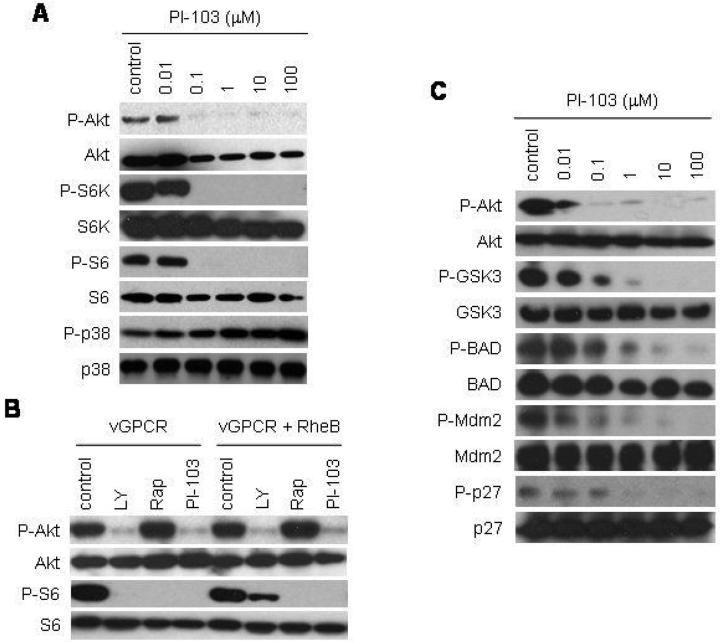
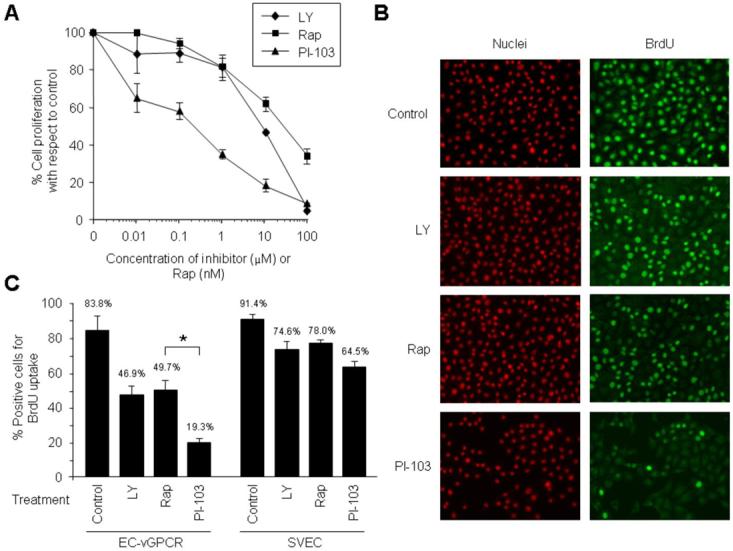
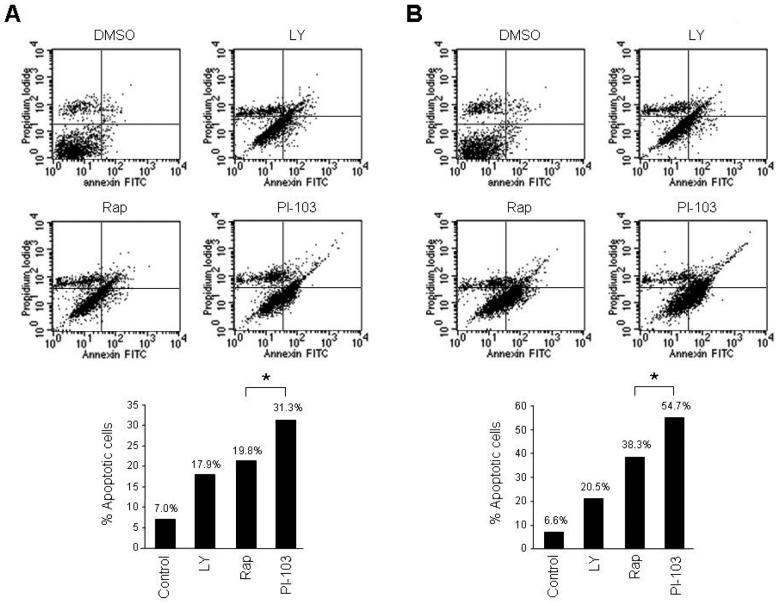
References
-
- Sodhi A, Montaner S, Gutkind JS. Does dysregulated expression of a deregulated viral GPCR trigger Kaposi's sarcomagenesis? Faseb J. 2004;18:422–7. - PubMed
-
- Sodhi A, Montaner S, Gutkind JS. Viral hijacking of G-protein-coupled-receptor signalling networks. Nat Rev Mol Cell Biol. 2004;5:998–1012. - PubMed
-
- Montaner S, Sodhi A, Molinolo A, et al. Endothelial infection with KSHV genes in vivo reveals that vGPCR initiates Kaposi's sarcomagenesis and can promote the tumorigenic potential of viral latent genes. Cancer Cell. 2003;3:23–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous
