

Rare codons cluster
- PMID: 18923675
- PMCID: PMC2565806
- DOI: 10.1371/journal.pone.0003412
Rare codons cluster
Abstract
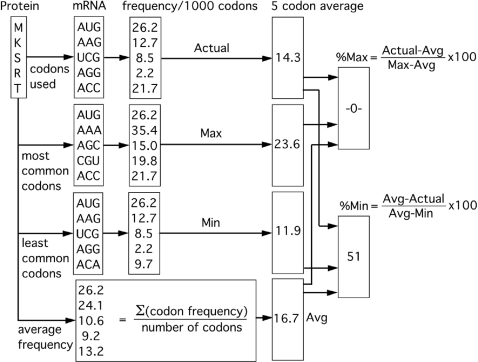
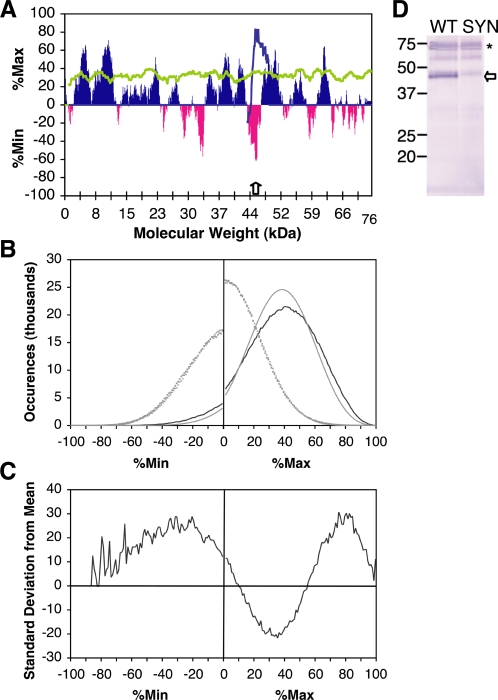
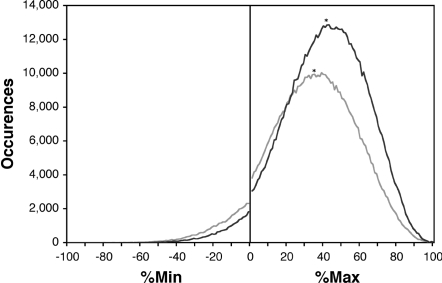
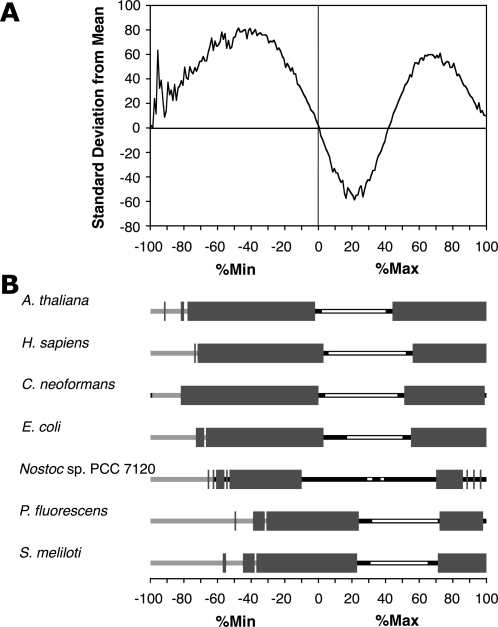
Most amino acids are encoded by more than one codon. These synonymous codons are not used with equal frequency: in every organism, some codons are used more commonly, while others are more rare. Though the encoded protein sequence is identical, selective pressures favor more common codons for enhanced translation speed and fidelity. However, rare codons persist, presumably due to neutral drift. Here, we determine whether other, unknown factors, beyond neutral drift, affect the selection and/or distribution of rare codons. We have developed a novel algorithm that evaluates the relative rareness of a nucleotide sequence used to produce a given protein sequence. We show that rare codons, rather than being randomly scattered across genes, often occur in large clusters. These clusters occur in numerous eukaryotic and prokaryotic genomes, and are not confined to unusual or rarely expressed genes: many highly expressed genes, including genes for ribosomal proteins, contain rare codon clusters. A rare codon cluster can impede ribosome translation of the rare codon sequence. These results indicate additional selective pressures govern the use of synonymous codons, and specifically that local pauses in translation can be beneficial for protein biogenesis.
Conflict of interest statement
Figures
References
-
- Duret L. Evolution of synonymous codon usage in metazoans. Curr Opin Genet Dev. 2002;12:640–649. - PubMed
-
- Kane JF. Effects of rare codon clusters on high-level expression of heterologous proteins in Escherichia coli. Curr Op Biotechnol. 1995;6:494–500. - PubMed
-
- Medigue C, Rouxel T, Vigier P, Henaut A, Danchin A. Evidence for horizontal gene transfer in Escherichia coli speciation. J Mol Biol. 1991;222:851–856. - PubMed
-
- Smith NG, Eyre-Walker A. Why are translationally sub-optimal synonymous codons used in Escherichia coli? J Mol Evol. 2001;53:225–236. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
