

Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease
- PMID: 18935984
- PMCID: PMC3268004
- DOI: 10.1016/j.dnarep.2008.09.004
Mitochondrial DNA damage is a hallmark of chemically induced and the R6/2 transgenic model of Huntington's disease
Abstract
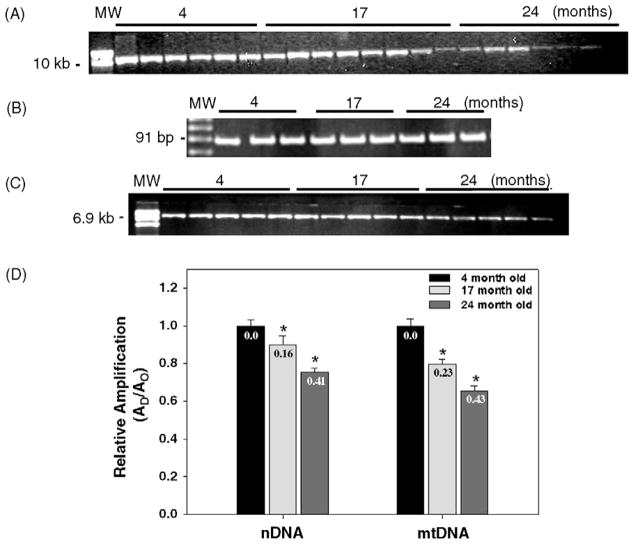
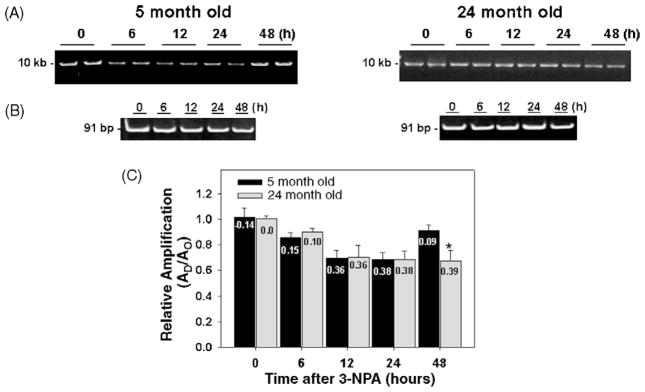
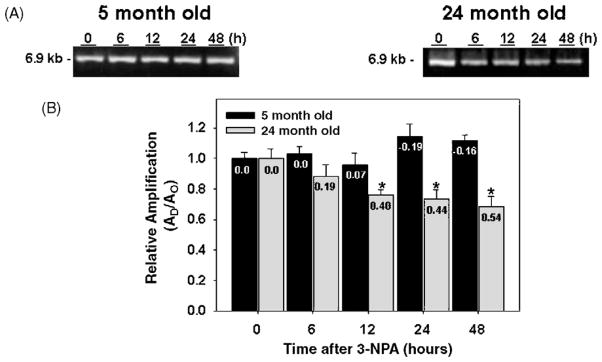
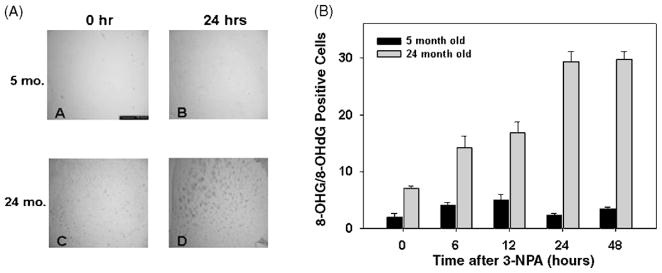
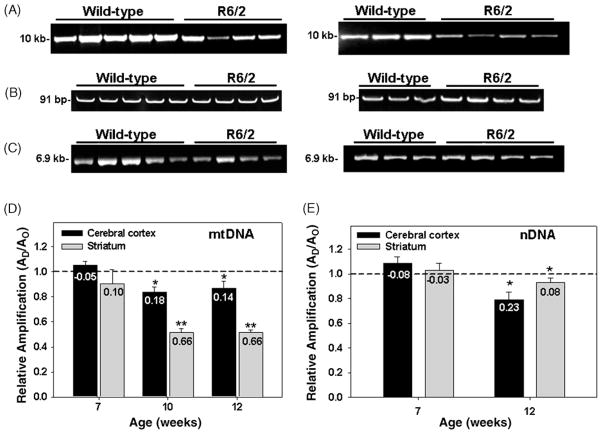
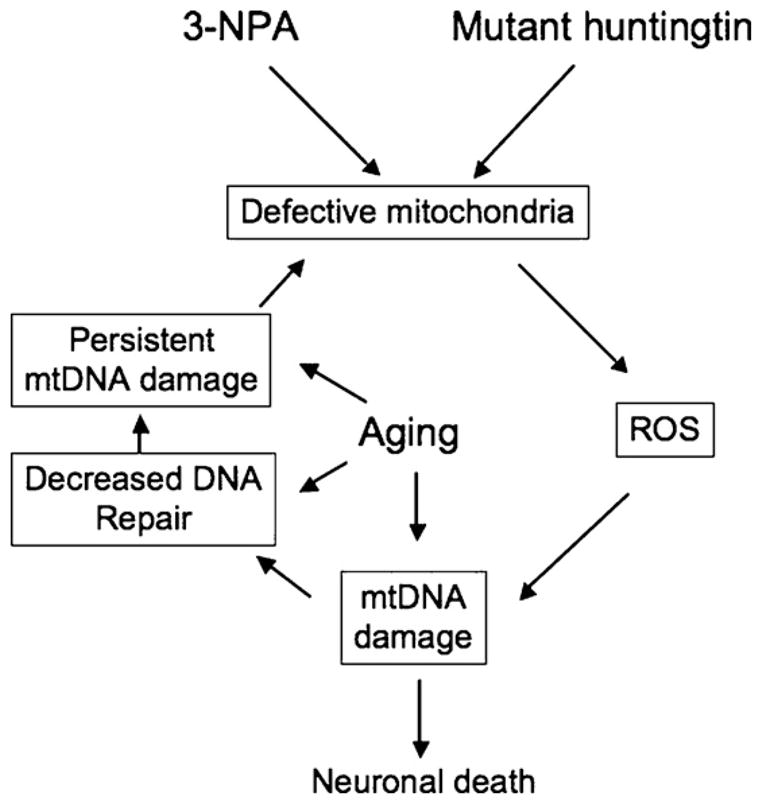
Many forms of neurodegeneration are associated with oxidative stress and mitochondrial dysfunction. Mitochondria are prominent targets of oxidative damage, however, it is not clear whether mitochondrial DNA (mtDNA) damage and/or its lack of repair are primary events in the delayed onset observed in Huntington's disease (HD). We hypothesize that an age-dependent increase in mtDNA damage contributes to mitochondrial dysfunction in HD. Two HD mouse models were studied, the 3-nitropropionic acid (3-NPA) chemically induced model and the HD transgenic mice of the R6/2 strain containing 115-150 CAG repeats in the huntingtin gene. The mitochondrial toxin 3-NPA inhibits complex II of the electron transport system and causes neurodegeneration that resembles HD in the striatum of human and experimental animals. We measured nuclear and mtDNA damage by quantitative PCR (QPCR) in striatum of 5- and 24-month-old untreated and 3-NPA treated C57BL/6 mice. Aging caused an increase in damage in both nuclear and mitochondrial genomes. 3-NPA induced 4-6 more damage in mtDNA than nuclear DNA in 5-month-old mice, and this damage was repaired by 48h in the mtDNA. In 24-month-old mice 3NPA caused equal amounts of nuclear and mitochondrial damage and this damage persistent in both genomes for 48h. QPCR analysis showed a progressive increase in the levels of mtDNA damage in the striatum and cerebral cortex of 7-12-week-old R6/2 mice. Striatum exhibited eight-fold more damage to the mtDNA compared with a nuclear gene. These data suggest that mtDNA damage is an early biomarker for HD-associated neurodegeneration and supports the hypothesis that mtDNA lesions may contribute to the pathogenesis observed in HD.
Conflict of interest statement
The authors declare no conflict of interest. The data included in this manuscript have not been published before.
Figures
References
-
- The Huntington’s Disease Collaborative Research Group. A novel gene encoding a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72:971–983. - PubMed
-
- Vonsattel JP, DiFiglia M. Huntington disease. J Neuropathol Exp Neurol. 1998;57:369–384. - PubMed
-
- Browne SE, Bowling AC, MacGarvey U, Baik MJ, Berger SC, Muqit MM, Bird ED, Beal MF. Oxidative damage and metabolic dysfunction in Huntington’s disease: selective vulnerability of the basal ganglia. Ann Neurol. 1997;41:646–653. - PubMed
-
- Polidori MC, Mecocci P, Browne SE, Senin U, Beal MF. Oxidative damage to mitochondrial DNA in Huntington’s disease parietal cortex. Neurosci Lett. 1999;272:53–56. - PubMed
-
- Bogdanov MB, Andreassen OA, Dedeoglu A, Ferrante RJ, Beal MF. Increased oxidative damage to DNA in a transgenic mouse model of Huntington’s disease. J Neurochem. 2001;79:1246–1249. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- NIGMS-R25-GM061838/PHS HHS/United States
- NCRR-2G12 RR030335-16/PHS HHS/United States
- R25 GM061838/GM/NIGMS NIH HHS/United States
- NCRR-G12RR03051/PHS HHS/United States
- G12 RR003051/RR/NCRR NIH HHS/United States
- NIGMS-S06 GM50695-08/PHS HHS/United States
- G12 MD007600/MD/NIMHD NIH HHS/United States
- S06 GM050695/GM/NIGMS NIH HHS/United States
- NINDS-U54 NS039408-06/NS/NINDS NIH HHS/United States
- U54 NS039408/NS/NINDS NIH HHS/United States
- G12 RR003035/RR/NCRR NIH HHS/United States
- NIA-R03 AG019015-01/AG/NIA NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
