

Audioprofile-directed screening identifies novel mutations in KCNQ4 causing hearing loss at the DFNA2 locus
- PMID: 18941426
- PMCID: PMC3337550
- DOI: 10.1097/GIM.0b013e318187e106
Audioprofile-directed screening identifies novel mutations in KCNQ4 causing hearing loss at the DFNA2 locus
Abstract
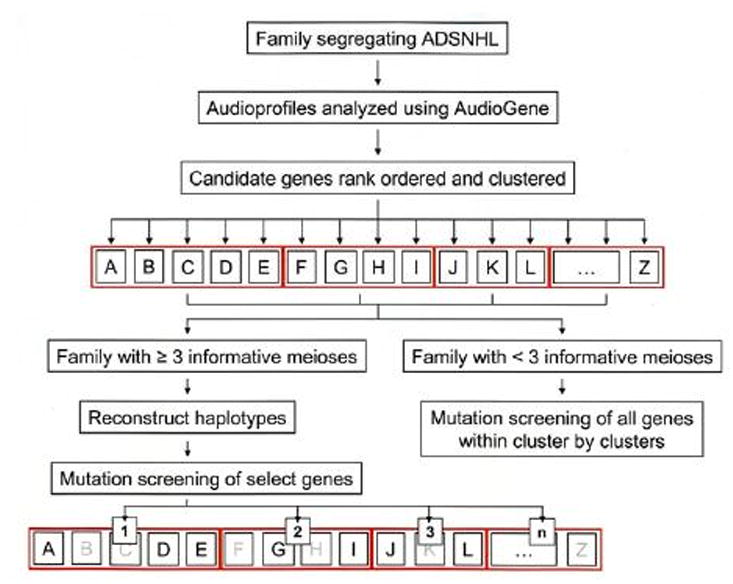
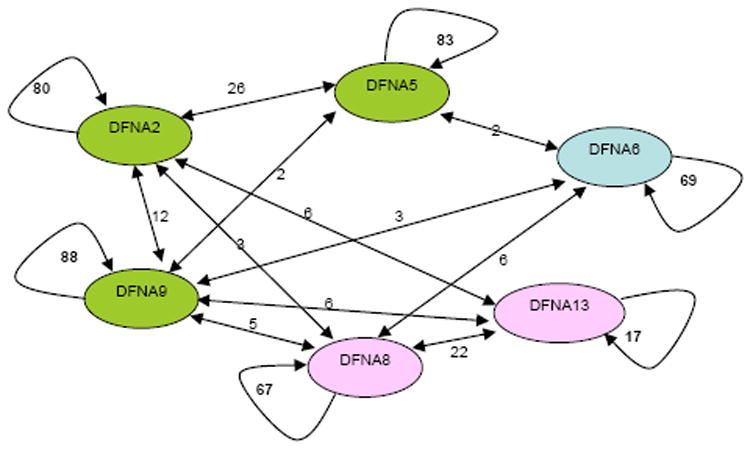
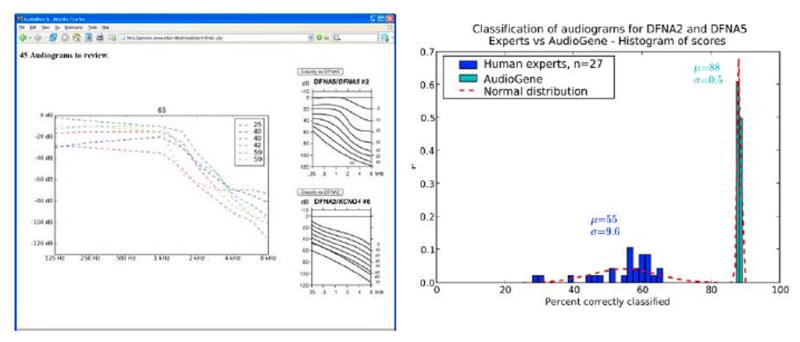
Purpose: Gene identification in small families segregating autosomal dominant sensorineural hearing loss presents a significant challenge. To address this challenge, we have developed a machine learning-based software tool, AudioGene v2.0, to prioritize candidate genes for mutation screening based on audioprofiling.
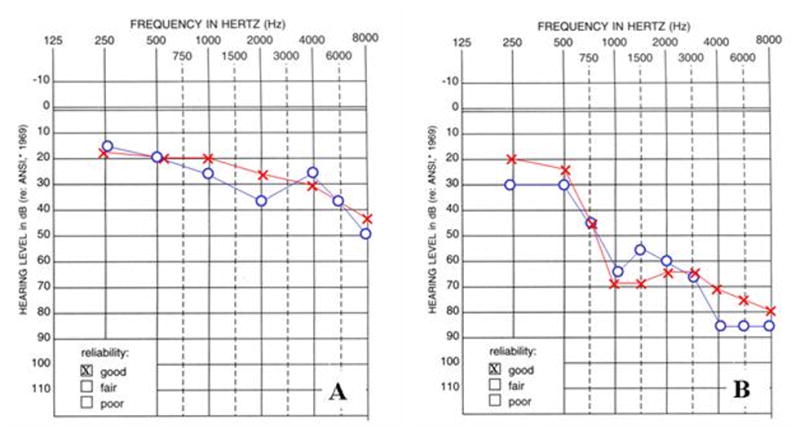
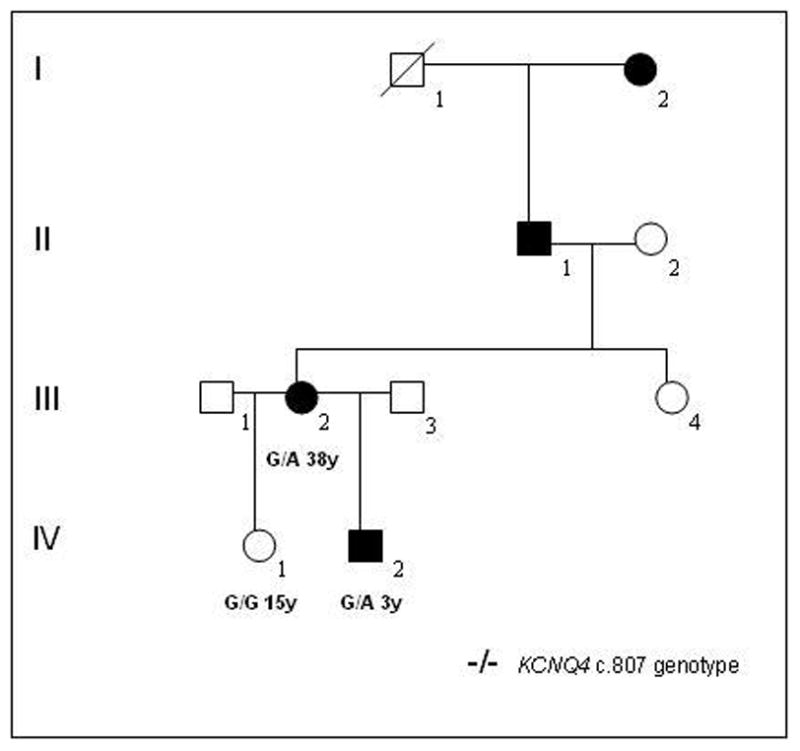
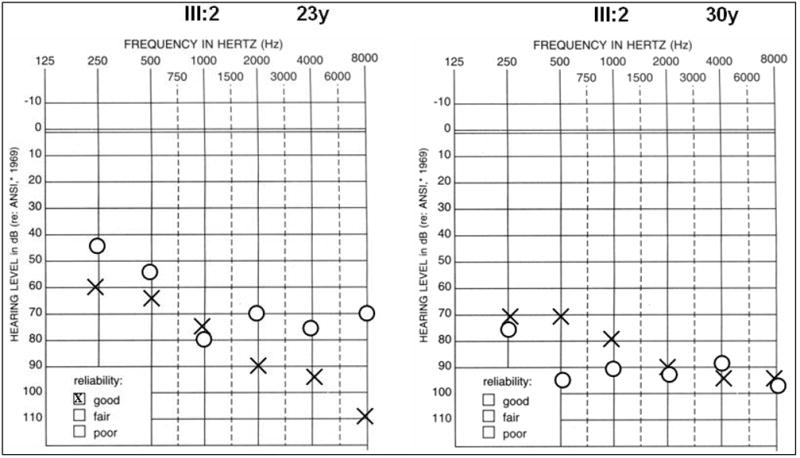
Methods: We analyzed audiometric data from a cohort of American families with high-frequency autosomal dominant sensorineural hearing loss. Those families predicted to have a DFNA2 audioprofile by AudioGene v2.0 were screened for mutations in the KCNQ4 gene.
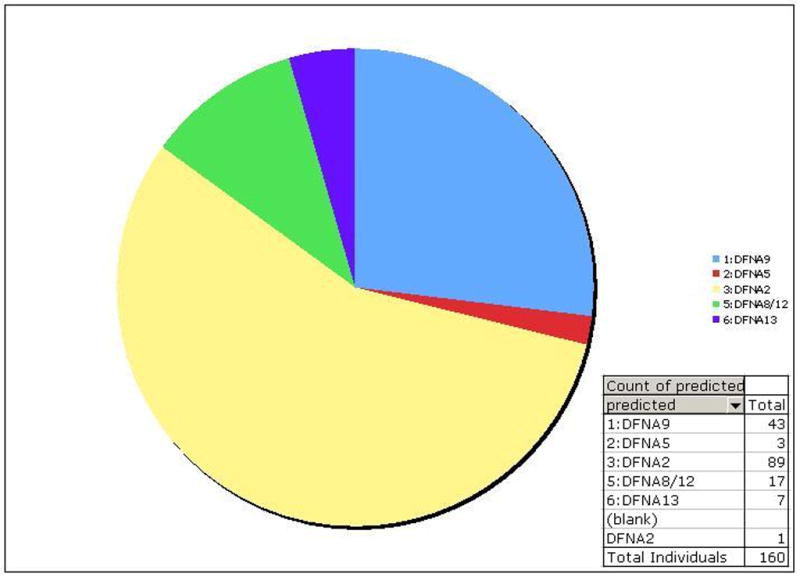
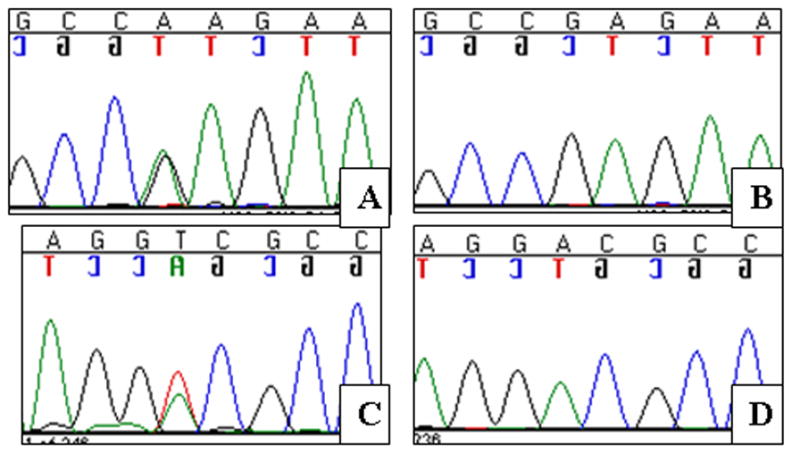
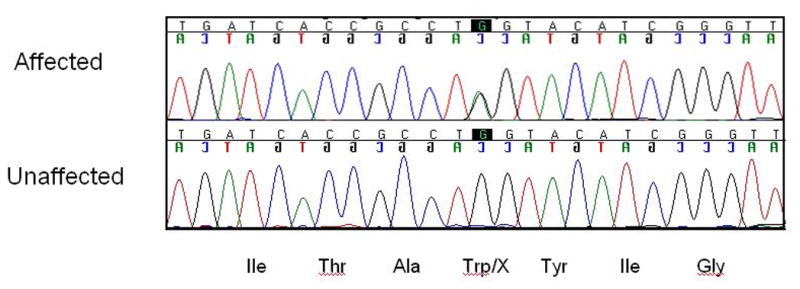
Results: Two novel missense mutations and a stop mutation were detected in three American families predicted to have DFNA2-related deafness for a positive predictive value of 6.3%. The false negative rate was 0%. The missense mutations were located in the channel pore region and the stop mutation was in transmembrane domain S5. The latter is the first DFNA2-causing stop mutation reported in KCNQ4.
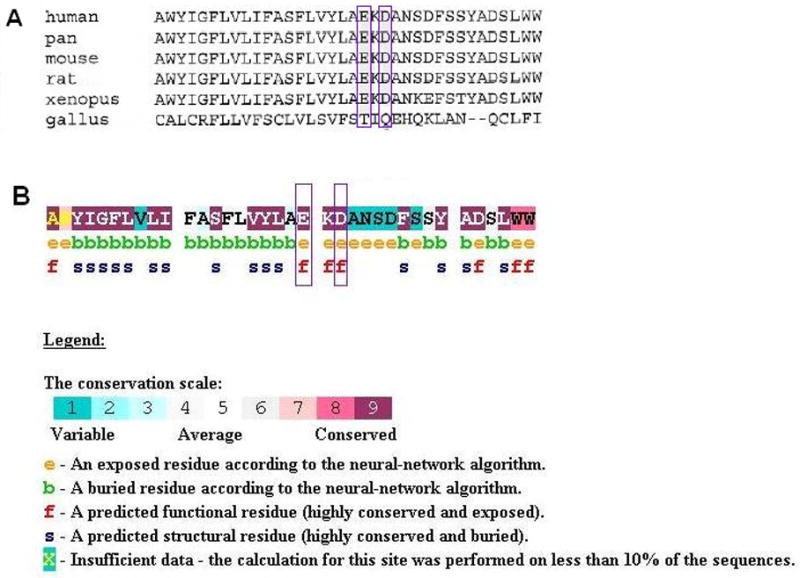
Conclusions: Our data suggest that the N-terminal end of the P-loop is crucial in maintaining the integrity of the KCNQ4 channel pore and AudioGene audioprofile analysis can effectively prioritize genes for mutation screening in small families segregating high-frequency autosomal dominant sensorineural hearing loss. AudioGene software will be made freely available to clinicians and researchers once it has been fully validated.
Conflict of interest statement
No researchers involved in this study report a conflict of interest.
Figures
References
-
- Morton NE. Genetic epidemiology of hearing impairment. Ann N Y Acad Sci. 1991;630:16–31. - PubMed
-
- Van Camp G, Smith RJ. Hereditary Hearing Loss Homepage. 2007.
-
- Huygen P, Pauw R, Cremers C. Genes, Hearing and Deafness. In: Martini M, Stephens D, Read AP, editors. From Molecular Biology to Clinical Practice - Audiometric profiles associated with genetic nonsyndromal hearing impairment: a review and phenotype analysis. London, UK: Informa Healthcare; 2007.
-
- Verhoeven K, Van Laer L, Kirschhofer K, et al. Mutations in the human alpha-tectorin gene cause autosomal dominant non-syndromic hearing impairment. Nat Genet. 1998;19(1):60–2. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
