

Complete genome viral phylogenies suggests the concerted evolution of regulatory cores and accessory satellites
- PMID: 18941535
- PMCID: PMC2567038
- DOI: 10.1371/journal.pone.0003500
Complete genome viral phylogenies suggests the concerted evolution of regulatory cores and accessory satellites
Abstract
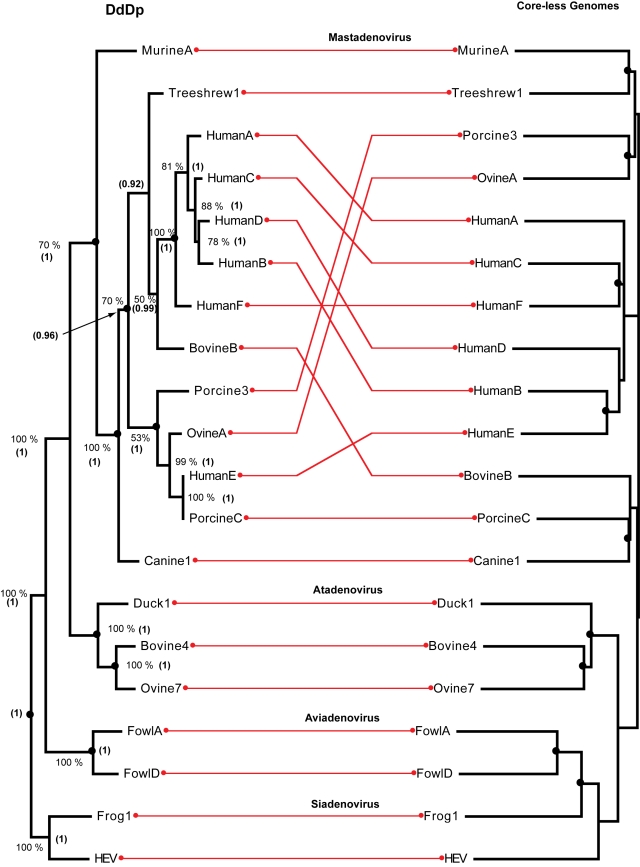
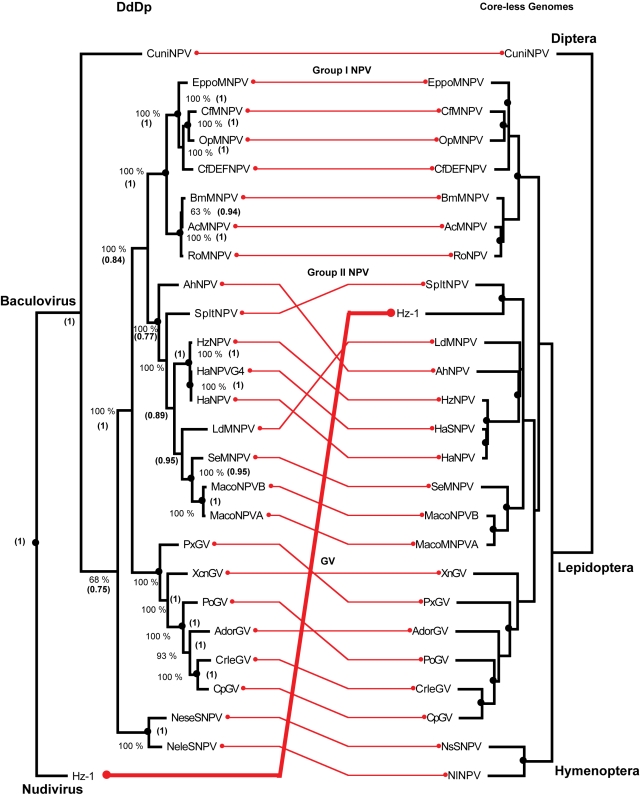
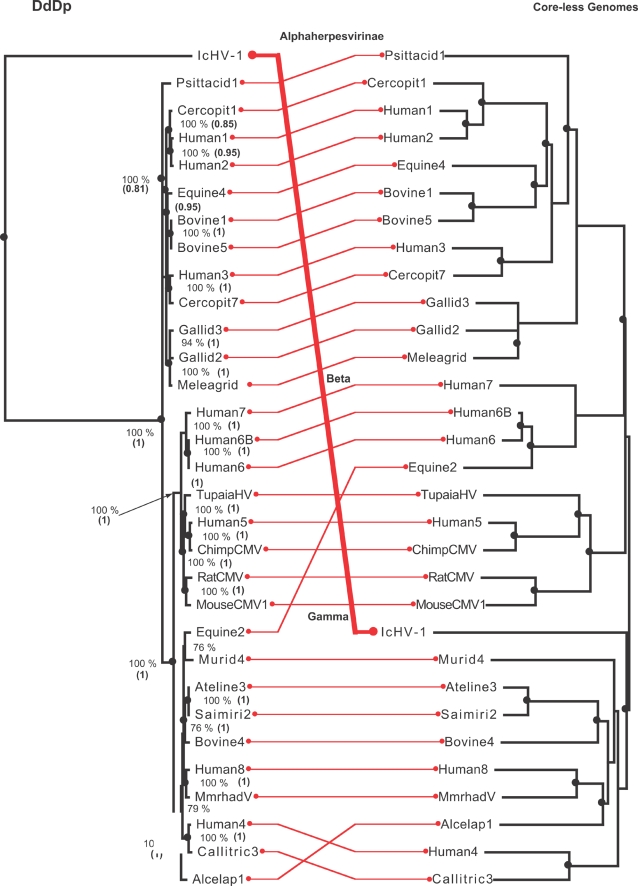
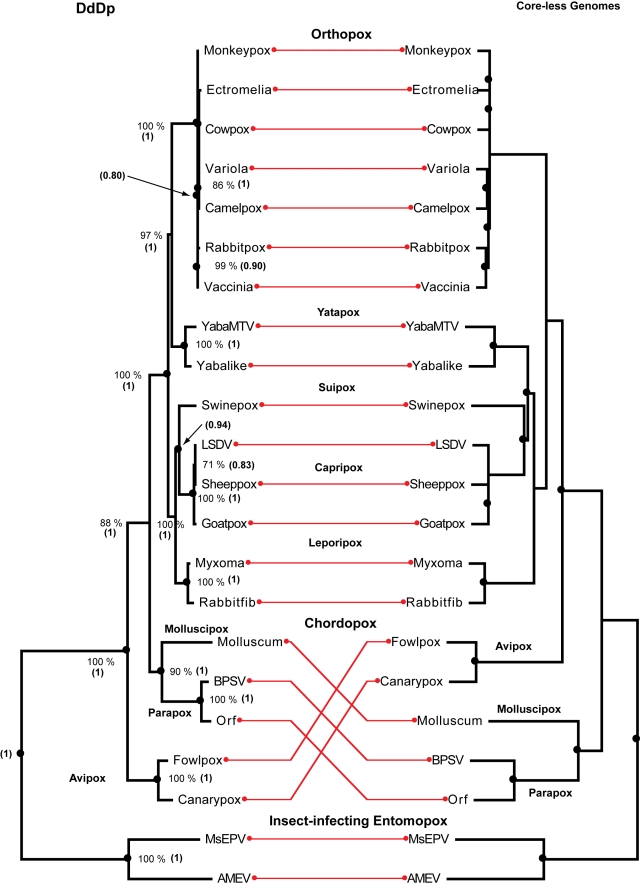
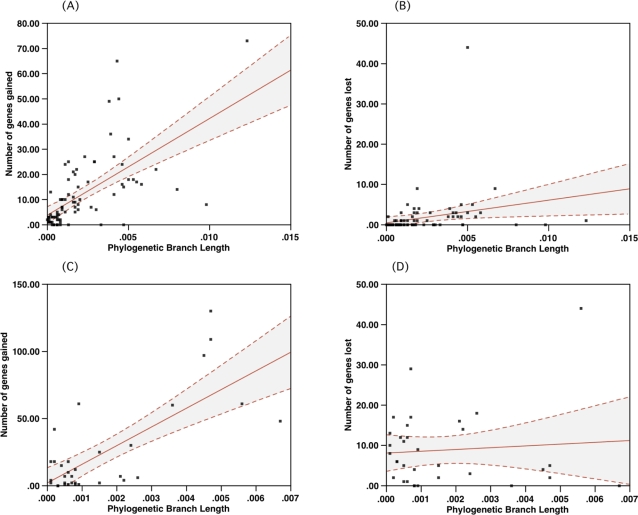
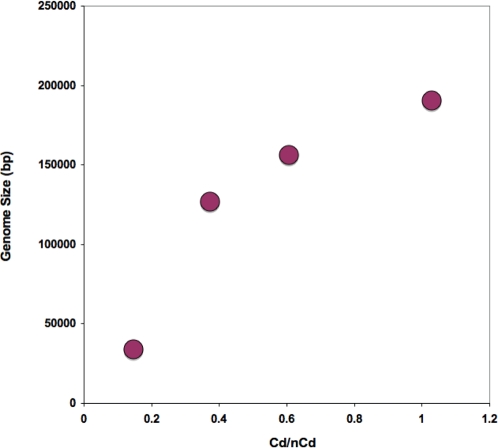
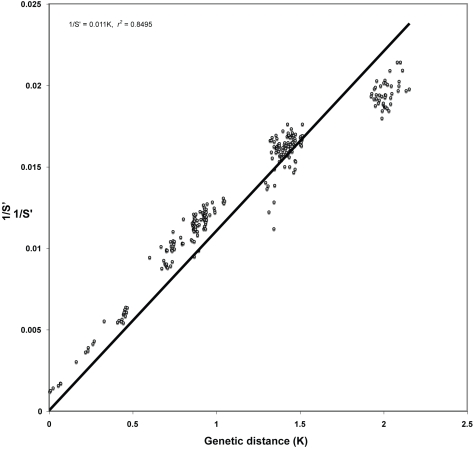
We consider the concerted evolution of viral genomes in four families of DNA viruses. Given the high rate of horizontal gene transfer among viruses and their hosts, it is an open question as to how representative particular genes are of the evolutionary history of the complete genome. To address the concerted evolution of viral genes, we compared genomic evolution across four distinct, extant viral families. For all four viral families we constructed DNA-dependent DNA polymerase-based (DdDp) phylogenies and in addition, whole genome sequence, as quantitative descriptions of inter-genome relationships. We found that the history of the polymerase gene was highly predictive of the history of the genome as a whole, which we explain in terms of repeated, co-divergence events of the core DdDp gene accompanied by a number of satellite, accessory genetic loci. We also found that the rate of gene gain in baculovirus and poxviruses proceeds significantly more quickly than the rate of gene loss and that there is convergent acquisition of satellite functions promoting contextual adaptation when distinct viral families infect related hosts. The congruence of the genome and polymerase trees suggests that a large set of viral genes, including polymerase, derive from a phylogenetically conserved core of genes of host origin, secondarily reinforced by gene acquisition from common hosts or co-infecting viruses within the host. A single viral genome can be thought of as a mutualistic network, with the core genes acting as an effective host and the satellite genes as effective symbionts. Larger virus genomes show a greater departure from linkage equilibrium between core and satellites functions.
Conflict of interest statement
Figures
References
-
- Herniou EA, Olszewski JA, Cory JS, O'Reilly DR. The genome sequence and evolution of baculovirus. Annu Rev Entomol. 2003;48:211–234. - PubMed
-
- Morse SS. Towards an evolutionary Biology of Viruses. In: Morse SS, editor. The Evolutionary Biology of Viruses. New York: Raven Press; 1994. pp. 1–27.
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
