

Origination of the split structure of spliceosomal genes from random genetic sequences
- PMID: 18941625
- PMCID: PMC2565106
- DOI: 10.1371/journal.pone.0003456
Origination of the split structure of spliceosomal genes from random genetic sequences
Abstract
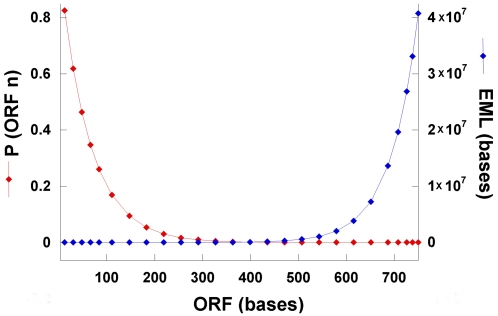
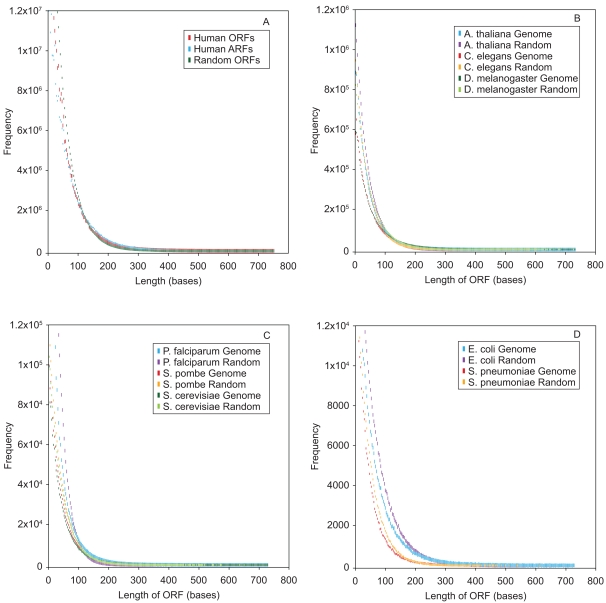
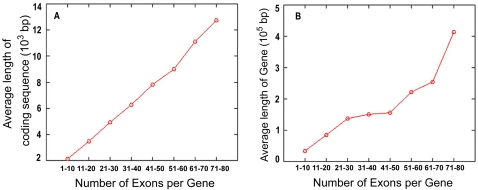
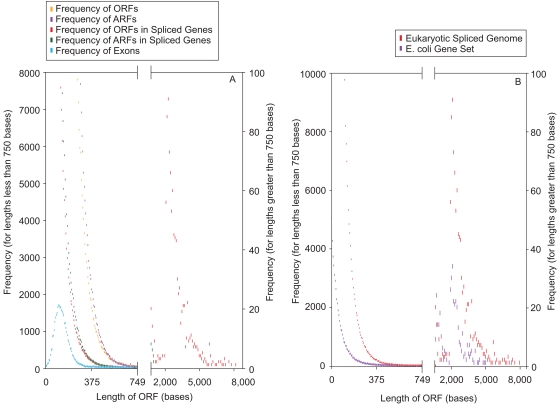
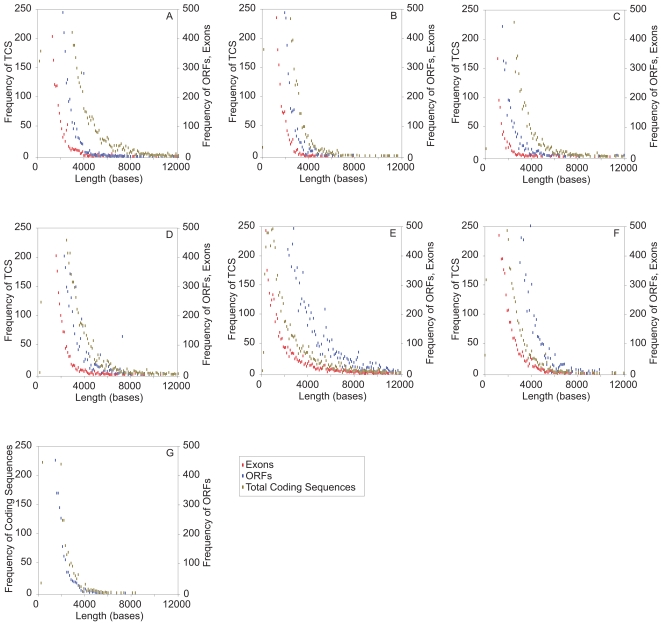
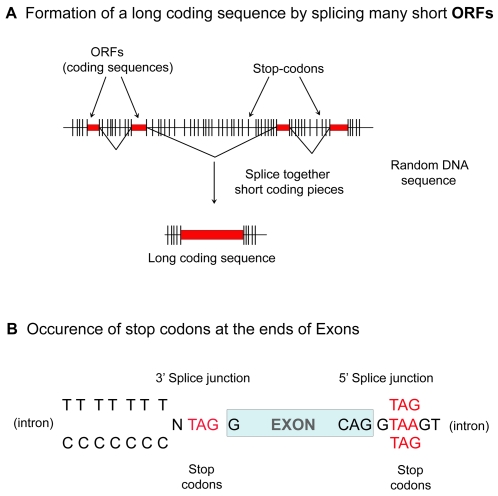
The mechanism by which protein-coding portions of eukaryotic genes came to be separated by long non-coding stretches of DNA, and the purpose for this perplexing arrangement, have remained unresolved fundamental biological problems for three decades. We report here a plausible solution to this problem based on analysis of open reading frame (ORF) length constraints in the genomes of nine diverse species. If primordial nucleic acid sequences were random in sequence, functional proteins that are innately long would not be encoded due to the frequent occurrence of stop codons. The best possible way that a long protein-coding sequence could have been derived was by evolving a split-structure from the random DNA (or RNA) sequence. Results of the systematic analyses of nine complete genome sequences presented here suggests that perhaps the major underlying structural features of split-genes have evolved due to the indigenous occurrence of split protein-coding genes in primordial random nucleotide sequence. The results also suggest that intron-rich genes containing short exons may have been the original form of genes intrinsically occurring in random DNA, and that intron-poor genes containing long exons were perhaps derived from the original intron-rich genes.
Conflict of interest statement
Figures

References
-
- Roy SW, Gilbert W. The evolution of spliceosomal introns: patterns, puzzles and progress. Nature Rev Genet. 2006;7:211–221. - PubMed
-
- Rogozin IB, Wolf YI, Sorokin AV, Mirkin BG, Koonin EV. Remarkable interkingdom conservation of intron positions and massive, lineage-specific intron loss and gain in eukaryotic evolution. Curr Biol. 2003;13:1512–1517. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
