

Placenta growth factor induces 5-lipoxygenase-activating protein to increase leukotriene formation in sickle cell disease
- PMID: 18945963
- PMCID: PMC2635078
- DOI: 10.1182/blood-2008-07-169821
Placenta growth factor induces 5-lipoxygenase-activating protein to increase leukotriene formation in sickle cell disease
Abstract
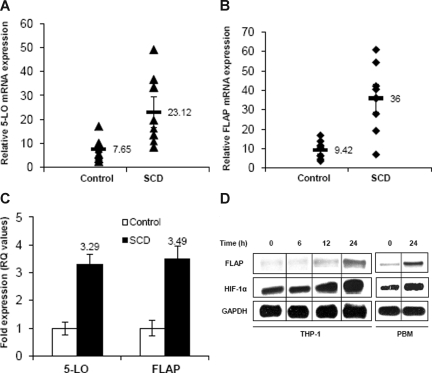
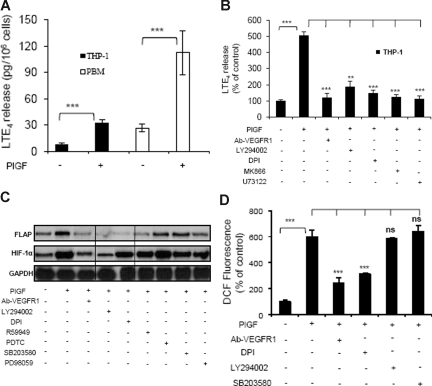
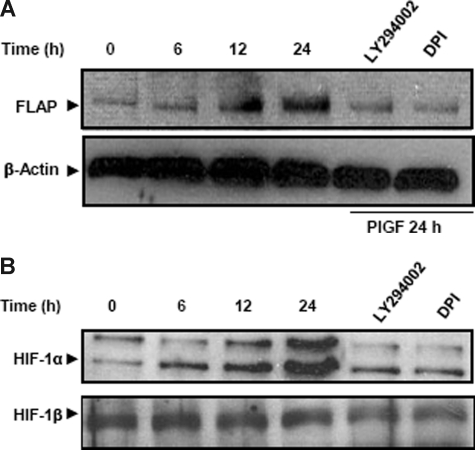
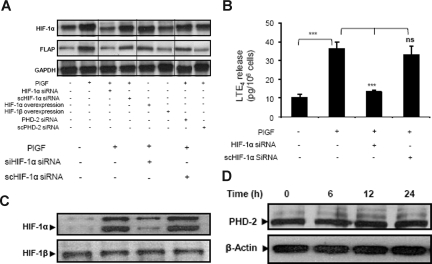
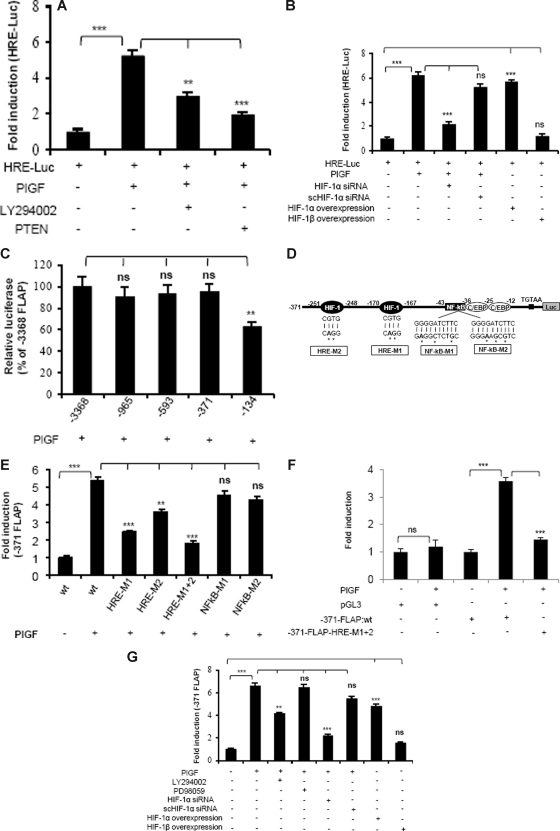
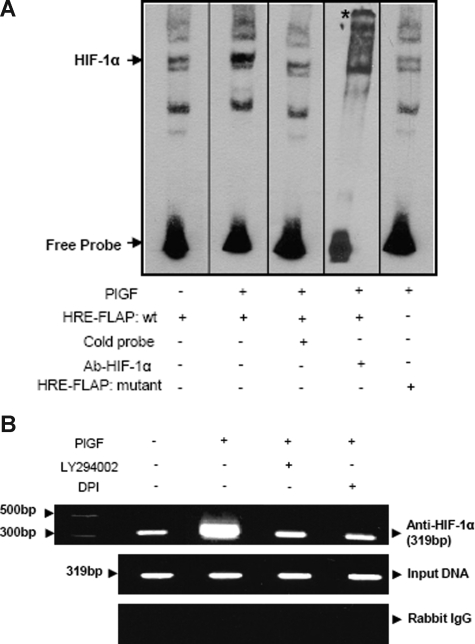
Individuals with sickle cell disease (SCD) have increased inflammation, a high incidence of airway hyperreactivity (AH), and increased circulating leukotrienes (LT). We show that expression of 5-lipoxygenase and 5-lipoxygenase activating protein (FLAP), key catalytic molecules in the LT pathway, were significantly increased in peripheral blood mononuclear cells (MNCs) in patients with SCD, compared with healthy controls. Placenta growth factor (PlGF), elaborated from erythroid cells, activated MNC and THP-1 monocytic cells to induce LT production. PlGF-mediated increased FLAP mRNA expression occurred via activation of phosphoinositide-3 (PI-3) kinase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, and hypoxia inducible factor-1alpha (HIF-1alpha). HIF-1alpha small interfering RNA (siRNA) reduced PlGF-induced FLAP expression. FLAP promoter-driven luciferase constructs demonstrated that PlGF-mediated luciferase induction was abrogated upon mutation of HIF-1alpha response element (HRE), but not the nuclear factor-kappaB (NF-kappaB) site in the FLAP promoter; a finding confirmed by chromatin immunoprecipitation (ChIP) analysis. PlGF also increased HIF-1alpha binding to the HRE in the FLAP promoter. Therefore, it is likely that the intrinsically elevated levels of PlGF in SCD subjects contribute to increased LT, which in turn, mediate both inflammation and AH. Herein, we identify a mechanism of increased LT in SCD and show HIF-1alpha as a hypoxia-independent target of PlGF. These studies provide new avenues to ameliorate these complications.
Figures
References
-
- Solovey A, Gui L, Ramakrishnan S, Steinberg MH, Hebbel RP. Sickle cell anemia as a possible state of enhanced anti-apoptotic tone: survival effect of vascular endothelial growth factor on circulating and unanchored endothelial cells. Blood. 1999;93:3824–3830. - PubMed
-
- Belcher JD, Marker PH, Weber JP, Hebbel RP, Vercellotti GM. Activated monocytes in sickle cell disease: potential role in the activation of vascular endothelium and vaso-occlusion. Blood. 2000;96:2451–2459. - PubMed
-
- Kaul DK, Liu XD, Choong S, et al. Anti-inflammatory therapy ameliorates leukocyte adhesion and microvascular flow abnormalities in transgenic sickle mice. Am J Physiol Heart Circ Physiol. 2004;287:H293–H301. - PubMed
-
- Croizat H, Nagel RL. Circulating cytokines response and the level of erythropoiesis in sickle cell anemia. Am J Hematol. 1999;60:105–115. - PubMed
-
- Miller ST, Sleeper LA, Pegelow CH, et al. Prediction of adverse outcomes in children with sickle cell disease. N Engl J Med. 2000;342:83–89. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
