

Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity
- PMID: 18948619
- PMCID: PMC2763388
- DOI: 10.1161/CIRCRESAHA.108.182832
Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity
Abstract
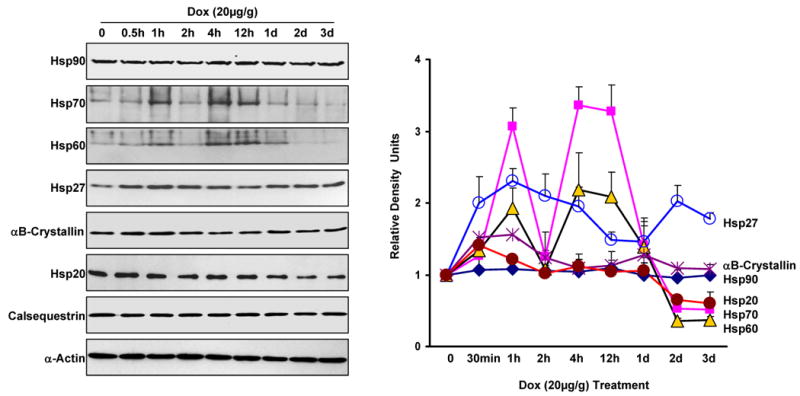
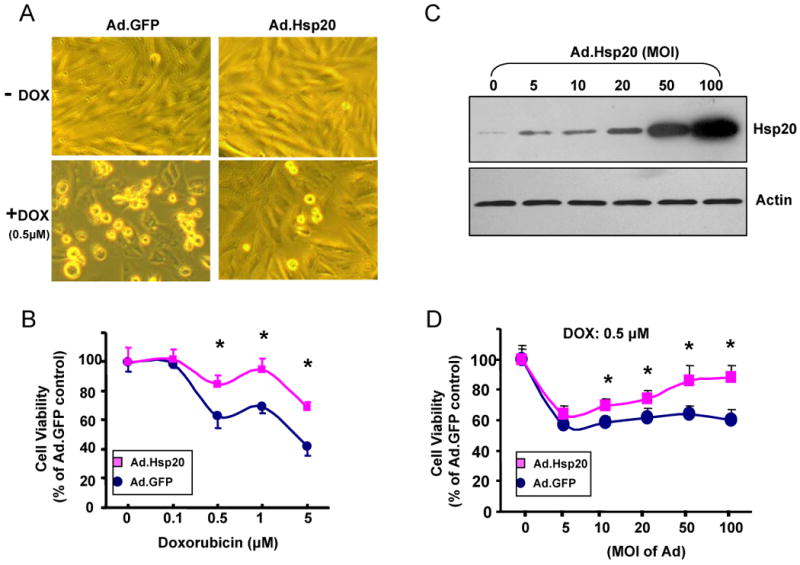
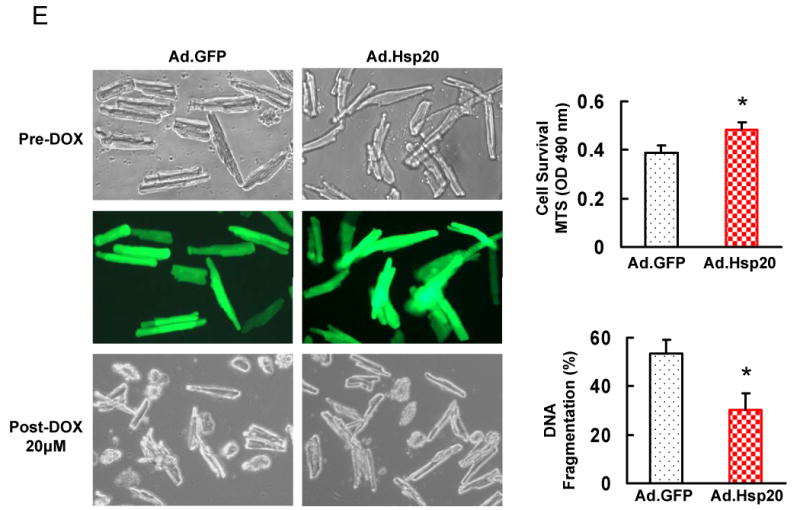
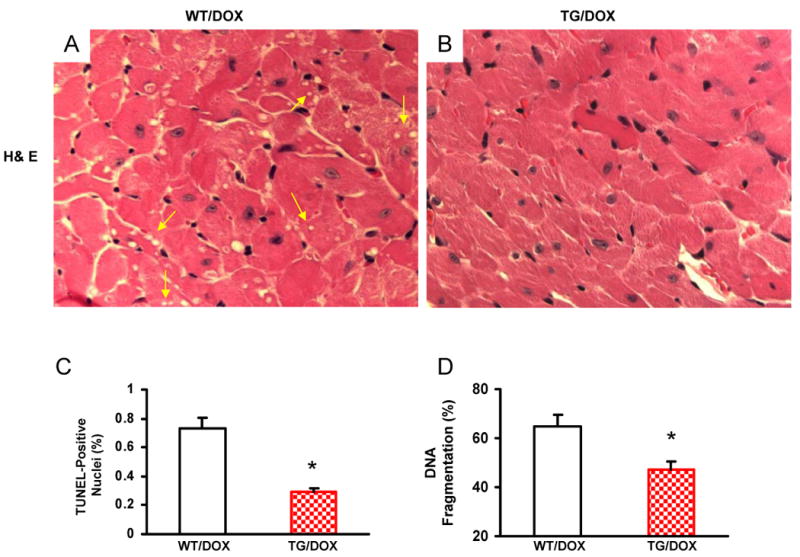
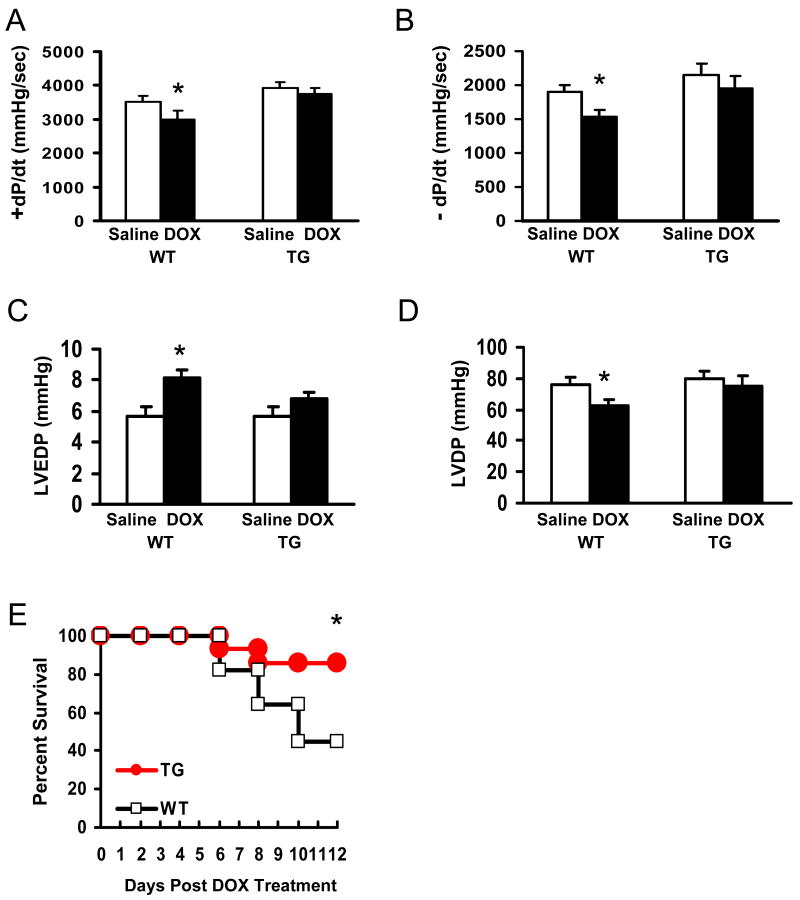
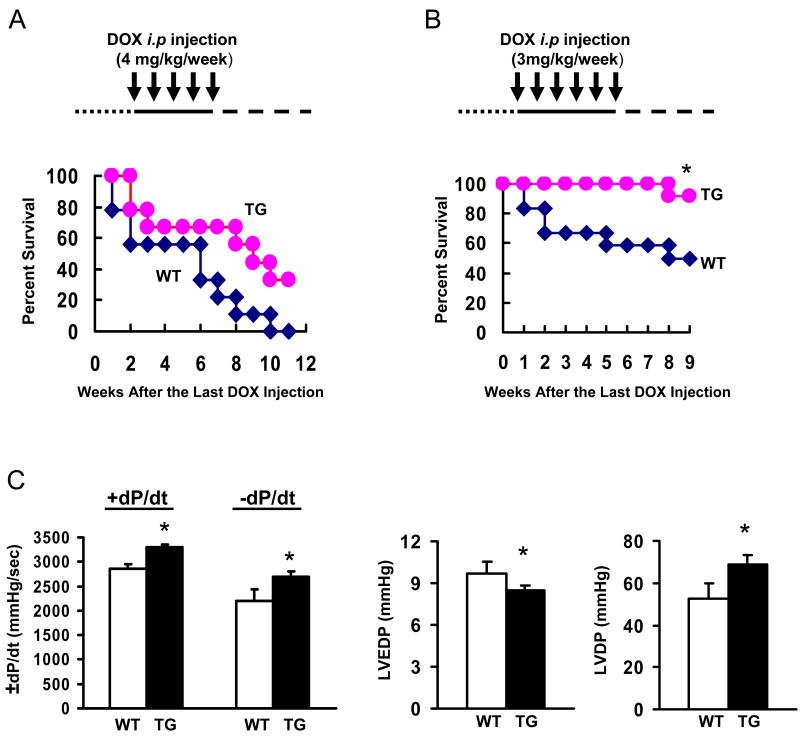
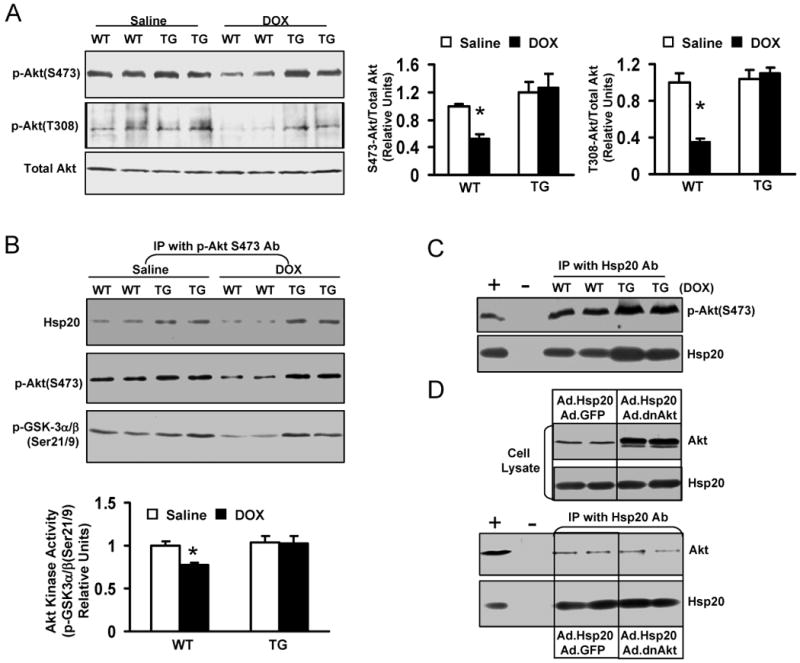
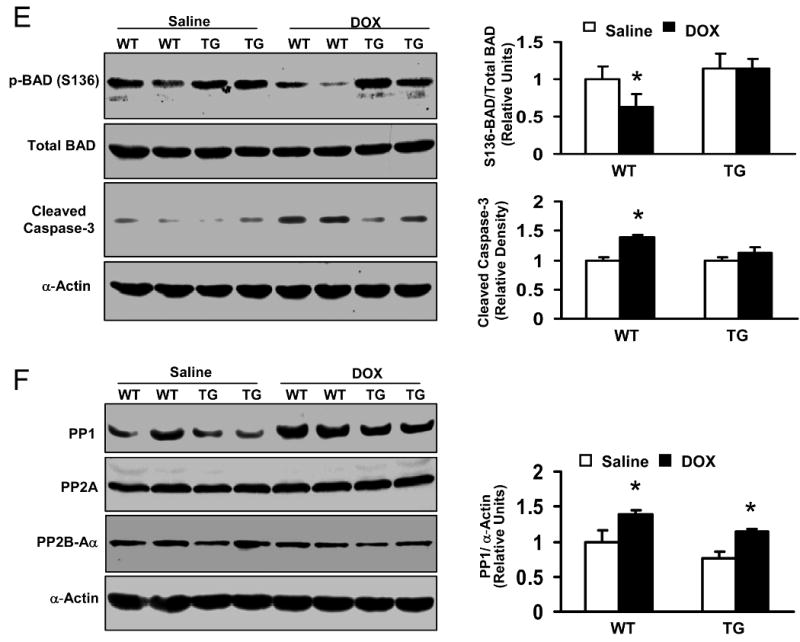
Doxorubicin (DOX) is a widely used antitumor drug, but its application is limited because of its cardiotoxic side effects. Heat shock protein (Hsp)20 has been recently shown to protect cardiomyocytes against apoptosis, induced by ischemia/reperfusion injury or by prolonged beta-agonist stimulation. However, it is not clear whether Hsp20 would exert similar protective effects against DOX-induced cardiac injury. Actually, DOX treatment was associated with downregulation of Hsp20 in the heart. To elucidate the role of Hsp20 in DOX-triggered cardiac toxicity, Hsp20 was first overexpressed ex vivo by adenovirus-mediated gene delivery. Increased Hsp20 levels conferred higher resistance to DOX-induced cell death, compared to green fluorescent protein control. Furthermore, cardiac-specific overexpression of Hsp20 in vivo significantly ameliorated acute DOX-triggered cardiomyocyte apoptosis and animal mortality. Hsp20 transgenic mice also showed improved cardiac function and prolonged survival after chronic administration of DOX. The mechanisms underlying these beneficial effects were associated with preserved Akt phosphorylation/activity and attenuation of DOX-induced oxidative stress. Coimmunoprecipitation studies revealed an interaction between Hsp20 and phosphorylated Akt. Accordingly, BAD phosphorylation was preserved, and cleaved caspase-3 was decreased in DOX-treated Hsp20 transgenic hearts, consistent with the antiapoptotic effects of Hsp20. Parallel ex vivo experiments showed that either infection with a dominant-negative Akt adenovirus or preincubation of cardiomyocytes with the phosphatidylinositol 3-kinase inhibitors significantly attenuated the protective effects of Hsp20. Taken together, our findings indicate that overexpression of Hsp20 inhibits DOX-triggered cardiac injury, and these beneficial effects appear to be dependent on Akt activation. Thus, Hsp20 may constitute a new therapeutic target in ameliorating the cardiotoxic effects of DOX treatment in cancer patients.
Figures
References
-
- Ganey PE, Carter LS, Mueller RA, Thurman RG. Doxorubicin toxicity in perfused rat heart. Decreased cell death at low oxygen tension. Circ Res. 1991;68:1610–1613. - PubMed
-
- Singal PK, Iliskovic N. Doxorubicin-induced cardiomyopathy. N Engl J Med. 1998;339:900–905. - PubMed
-
- Safra T, Muggia F, Jeffers S, Tsao-Wei DD, Groshen S, Lyass O, Henderson R, Berry G, Gabizon A. Pegylated liposomal doxorubicin (doxil): reduced clinical cardiotoxicity in patients reaching or exceeding cumulative doses of 500 mg/m2. Ann Oncol. 2000;11:1029–1033. - PubMed
-
- Kluza J, Marchetti P, Gallego MA, Lancel S, Fournier C, Loyens A, Beauvillain JC, Bailly C. Mitochondrial proliferation during apoptosis induced by anticancer agents: effects of doxorubicin and mitoxantrone on cancer and cardiac cells. Oncogene. 2004;23:7018–7030. - PubMed
-
- Wang S, Konorev EA, Kotamraju S, Joseph J, Kalivendi S, Kalyanaraman B. Doxorubicin induces apoptosis in normal and tumor cells via distinctly different mechanisms. J Biol Chem. 2004;279:25535–25543. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
