

Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications
- PMID: 18951461
- PMCID: PMC3938023
- DOI: 10.1002/humu.20896
Classification of rare missense substitutions, using risk surfaces, with genetic- and molecular-epidemiology applications
Abstract
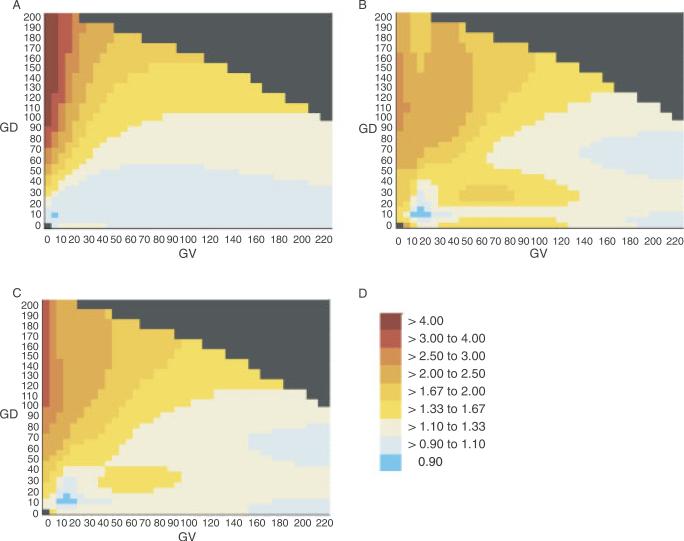
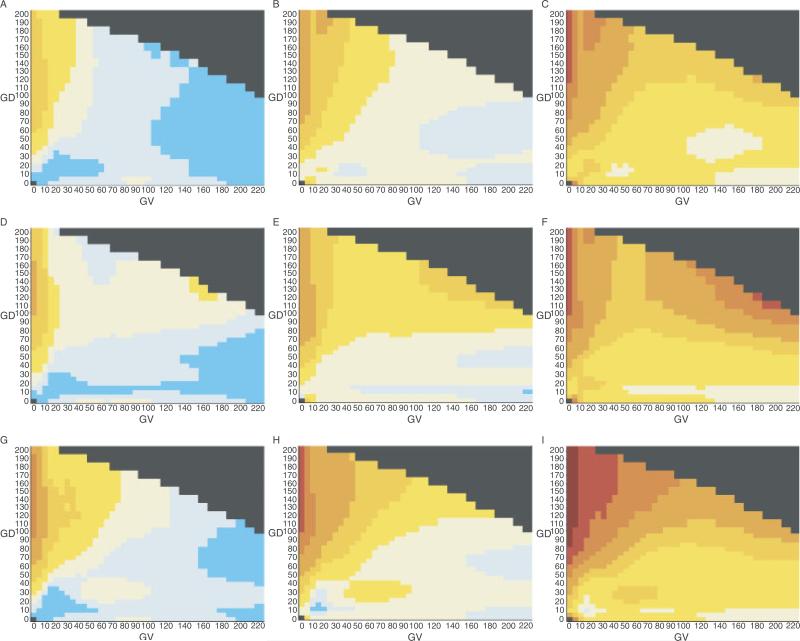
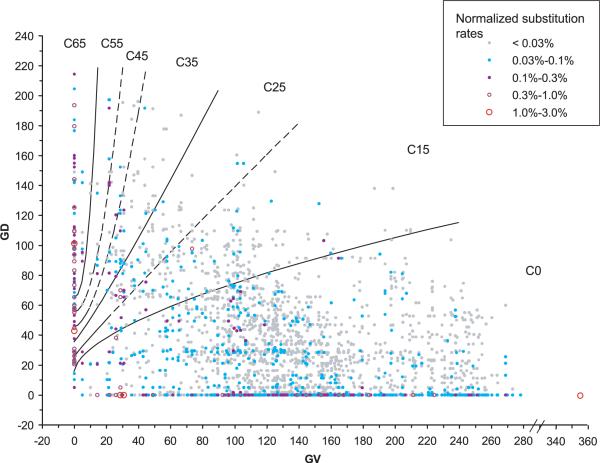
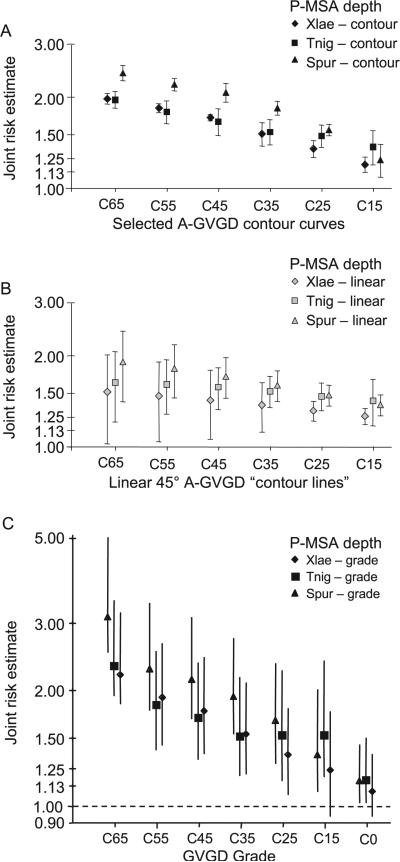
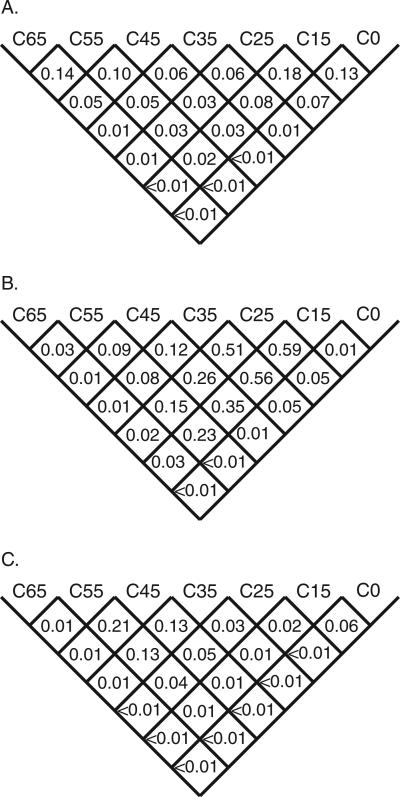
Many individually rare missense substitutions are encountered during deep resequencing of candidate susceptibility genes and clinical mutation screening of known susceptibility genes. BRCA1 and BRCA2 are among the most resequenced of all genes, and clinical mutation screening of these genes provides an extensive data set for analysis of rare missense substitutions. Align-GVGD is a mathematically simple missense substitution analysis algorithm, based on the Grantham difference, which has already contributed to classification of missense substitutions in BRCA1, BRCA2, and CHEK2. However, the distribution of genetic risk as a function of Align-GVGD's output variables Grantham variation (GV) and Grantham deviation (GD) has not been well characterized. Here, we used data from the Myriad Genetic Laboratories database of nearly 70,000 full-sequence tests plus two risk estimates, one approximating the odds ratio and the other reflecting strength of selection, to display the distribution of risk in the GV-GD plane as a series of surfaces. We abstracted contours from the surfaces and used the contours to define a sequence of missense substitution grades ordered from greatest risk to least risk. The grades were validated internally using a third, personal and family history-based, measure of risk. The Align-GVGD grades defined here are applicable to both the genetic epidemiology problem of classifying rare missense substitutions observed in known susceptibility genes and the molecular epidemiology problem of analyzing rare missense substitutions observed during case-control mutation screening studies of candidate susceptibility genes.
(c) 2008 Wiley-Liss, Inc.
Figures
References
-
- Capriotti E, Arbiza L, Casadio R, Dopazo J, Dopazo H, Marti-Renom MA. Use of estimated evolutionary strength at the codon level improves the prediction of disease-related protein mutations in humans. Hum Mutat. 2008;29:198–204. - PubMed
-
- Chan PA, Duraisamy S, Miller PJ, Newell JA, McBride C, Bond JP, Raevaara T, Ollila S, Nystrom M, Grimm AJ, Christodoulou J, Oetting WS, Greenblatt MS. Interpreting missense variants: comparing computational methods in human disease genes CDKN2A, MLH1, MSH2, MECP2, and tyrosinase (TYR). Hum Mutat. 2007;28:683–693. - PubMed
-
- Chenevix-Trench G, Healey S, Lakhani S, Waring P, Cummings M, Brinkworth R, Deffenbaugh AM, Burbidge LA, Pruss D, Judkins T, Scholl T, Bekessy A, Marsh A, Lovelock P, Wong M, Tesoriero A, Renard H, Southey M, Hopper JL, Yannoukakos K, Brown M, Easton D, Tavtigian SV, Goldgar D, Spurdle AB. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006;66:2019–2027. - PubMed
-
- Cohen JC, Kiss RS, Pertsemlidis A, Marcel YL, McPherson R, Hobbs HH. Multiple rare alleles contribute to low plasma levels of HDL cholesterol. Science. 2004;305:869–872. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
