

Mutant huntingtin and mitochondrial dysfunction
- PMID: 18951640
- PMCID: PMC2613540
- DOI: 10.1016/j.tins.2008.09.004
Mutant huntingtin and mitochondrial dysfunction
Abstract
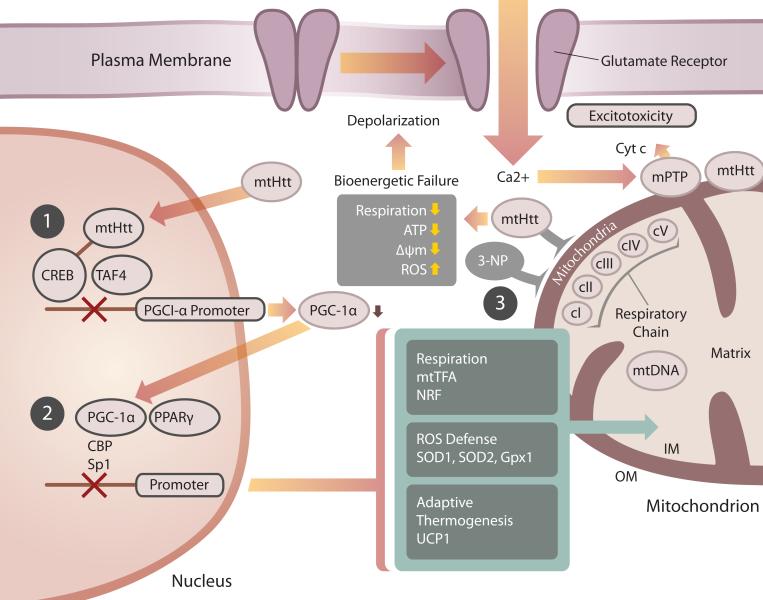
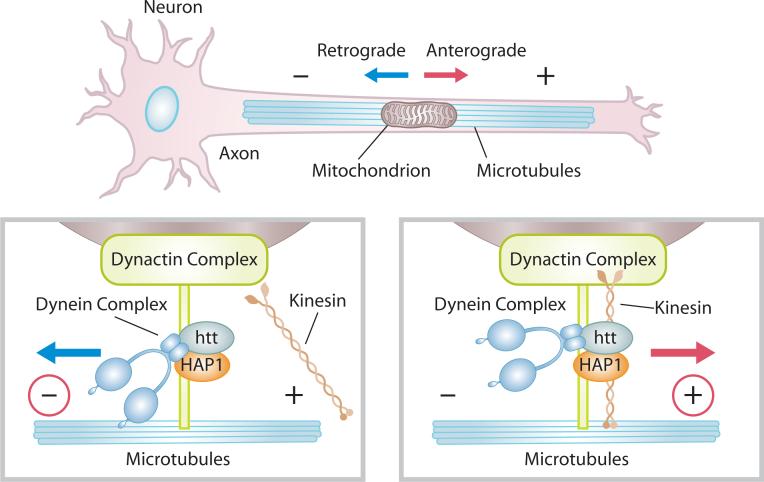
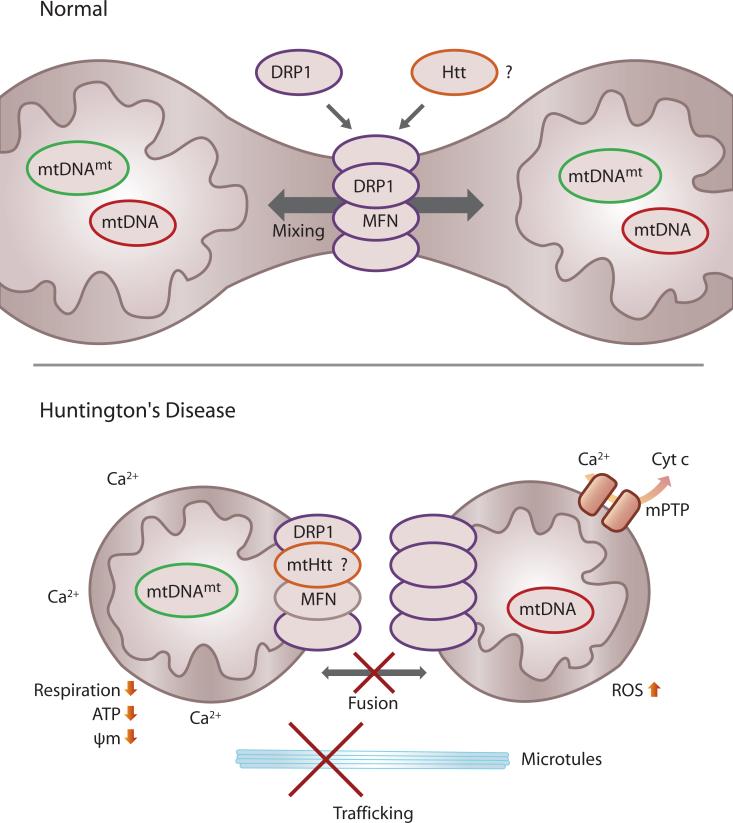
Huntington's disease (HD) is a fatal, inherited neurodegenerative disorder that gradually robs affected individuals of memory, cognitive skills and normal movements. Although research has identified a single faulty gene, the huntingtin gene, as the cause of the disease, a cure remains elusive. Strong evidence indicates that mitochondrial impairment plays a key part in HD pathogenesis. Here, we highlight how mutant huntingtin (mtHtt) might cause mitochondrial dysfunction by either perturbing transcription of nuclear-encoded mitochondrial proteins or by direct interaction with the organelle and modulation of respiration, mitochondrial membrane potential and Ca(2+) buffering. In addition, we propose that mtHtt might convey its neurotoxicity by evoking defects in mitochondrial dynamics, organelle trafficking and fission and fusion, which, in turn, might result in bioenergetic failure and HD-linked neuronal dysfunction and cell death. Finally, we speculate how mitochondria might dictate selective vulnerability of long projection neurons, such as medium spiny neurons, which are particularly affected in HD.
Figures
References
-
- Goldberg YP, et al. Identification of an Alu retrotransposition event in close proximity to a strong candidate gene for Huntington's disease. Nature. 1993;362:370–373. - PubMed
-
- A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington's disease chromosomes. The Huntington's Disease Collaborative Research Group. Cell. 1993;72:971–983. - PubMed
-
- Kegel KB, et al. Huntingtin is present in the nucleus, interacts with the transcriptional corepressor C-terminal binding protein, and represses transcription. J Biol Chem. 2002;277:7466–7476. - PubMed
-
- Kegel KB, et al. Huntingtin associates with acidic phospholipids at the plasma membrane. J Biol Chem. 2005;280:36464–36473. - PubMed
-
- Rockabrand E, et al. The first 17 amino acids of Huntingtin modulate its sub-cellular localization, aggregation and effects on calcium homeostasis. Hum Mol Genet. 2007;16:61–77. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
