

Principal component analysis for protein folding dynamics
- PMID: 18952103
- PMCID: PMC2652707
- DOI: 10.1016/j.jmb.2008.10.018
Principal component analysis for protein folding dynamics
Abstract
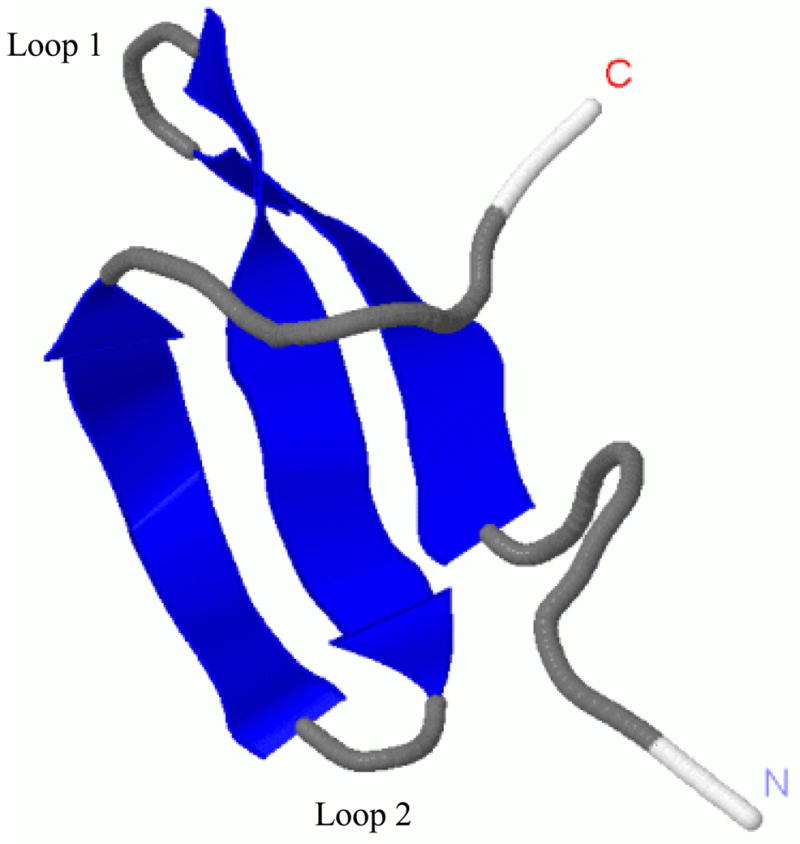
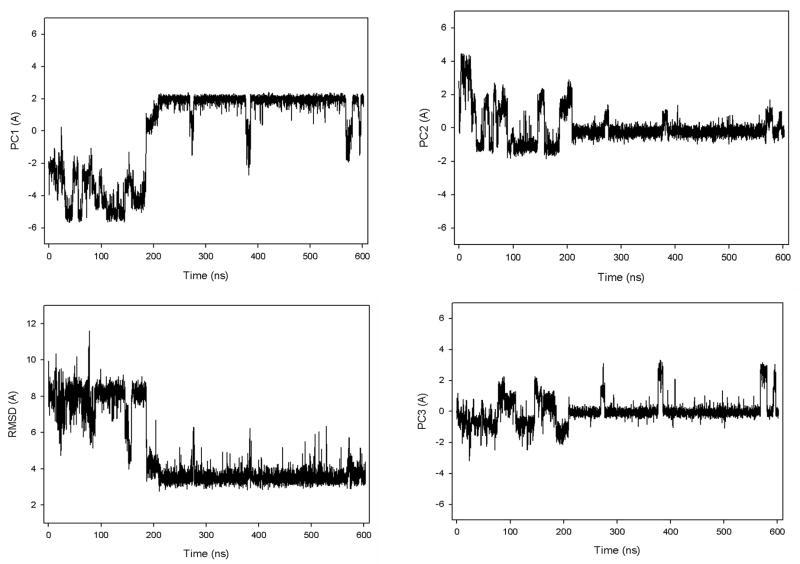
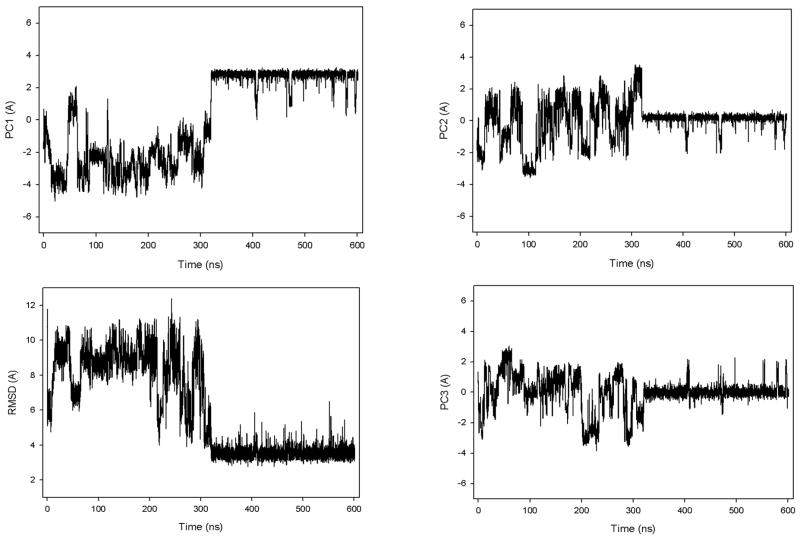
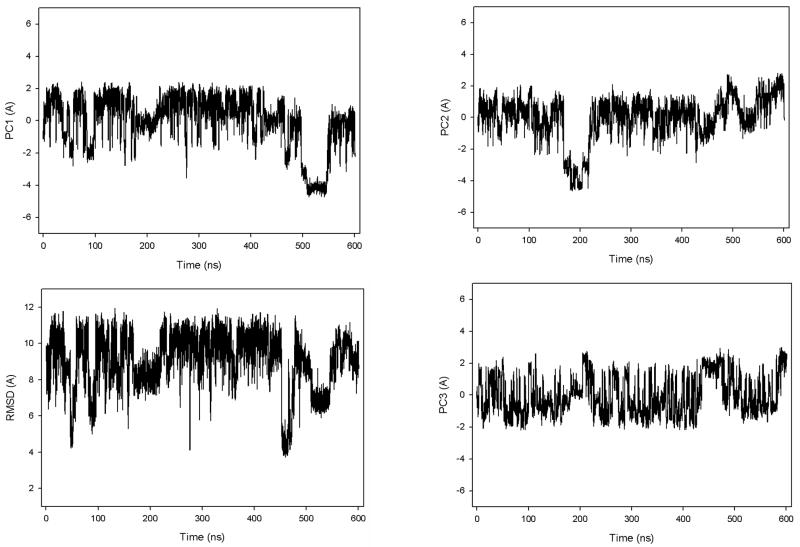
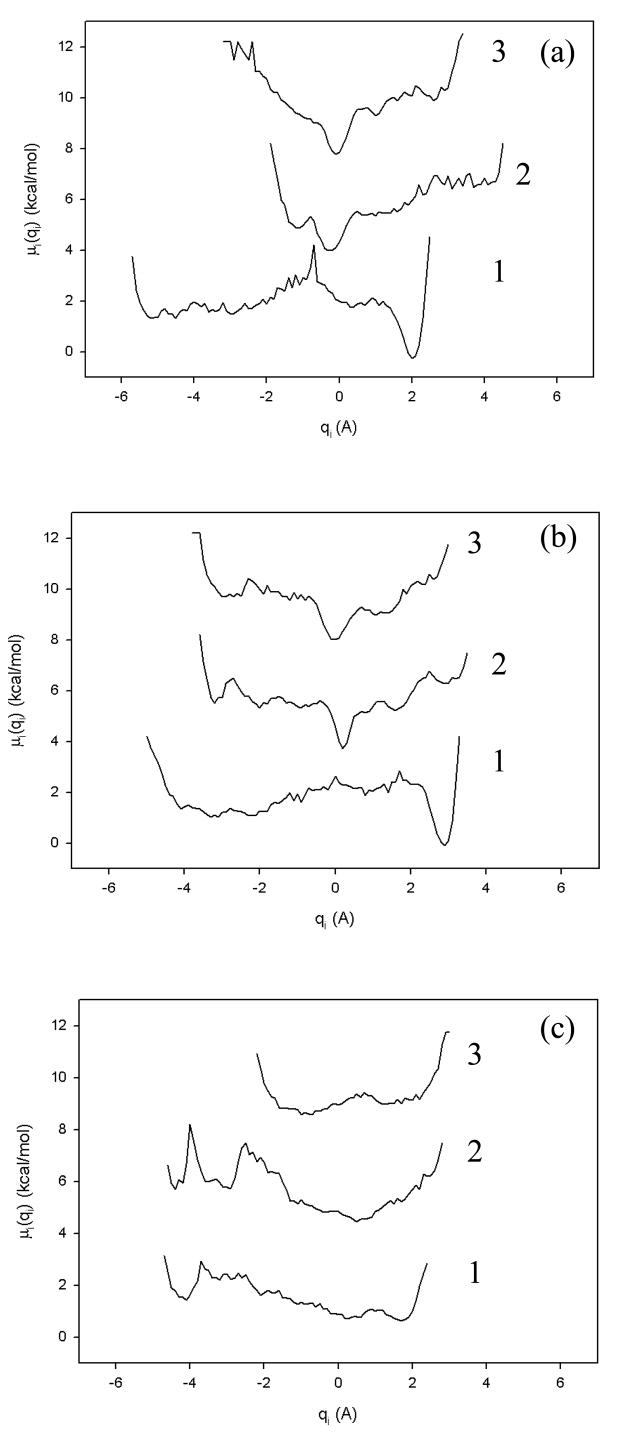
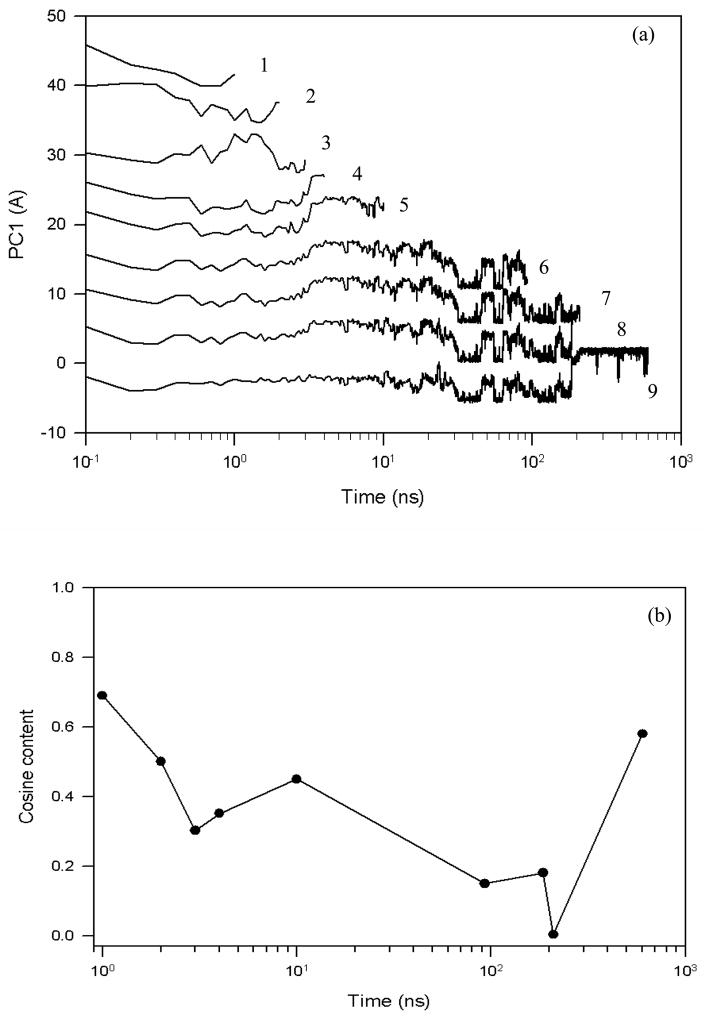
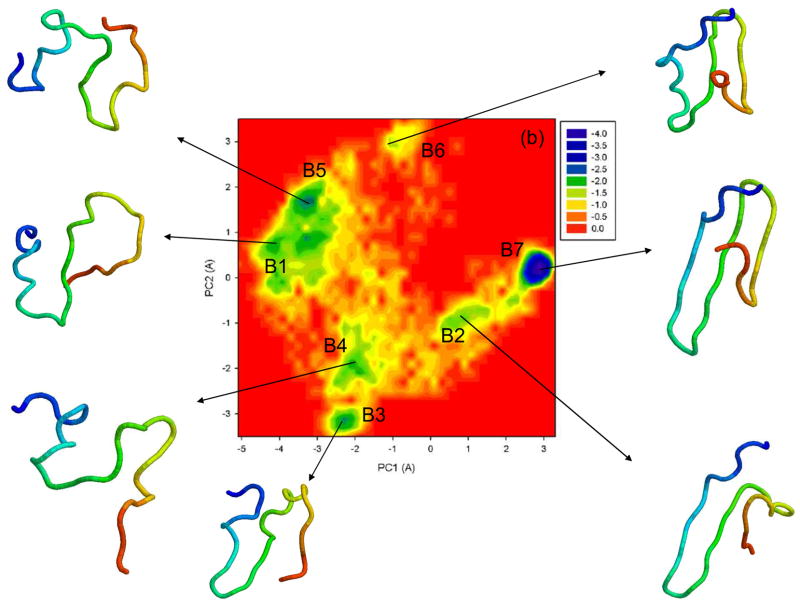
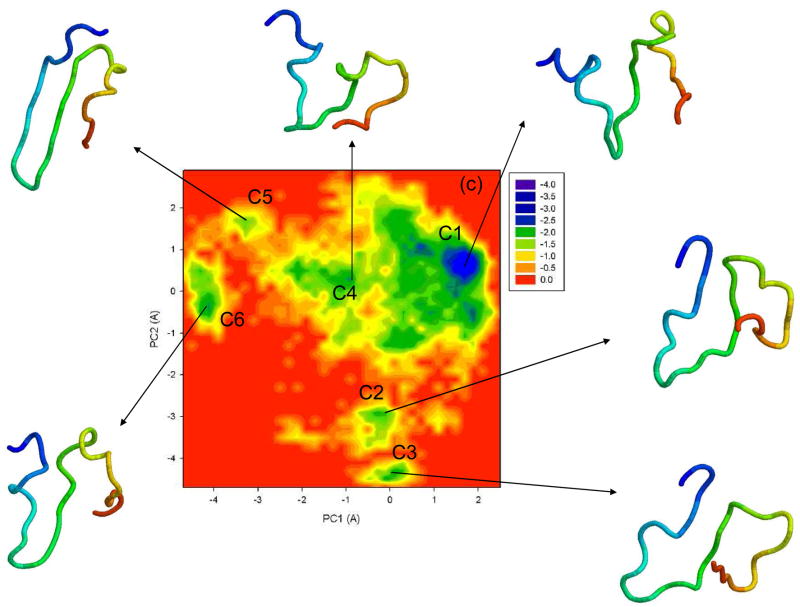
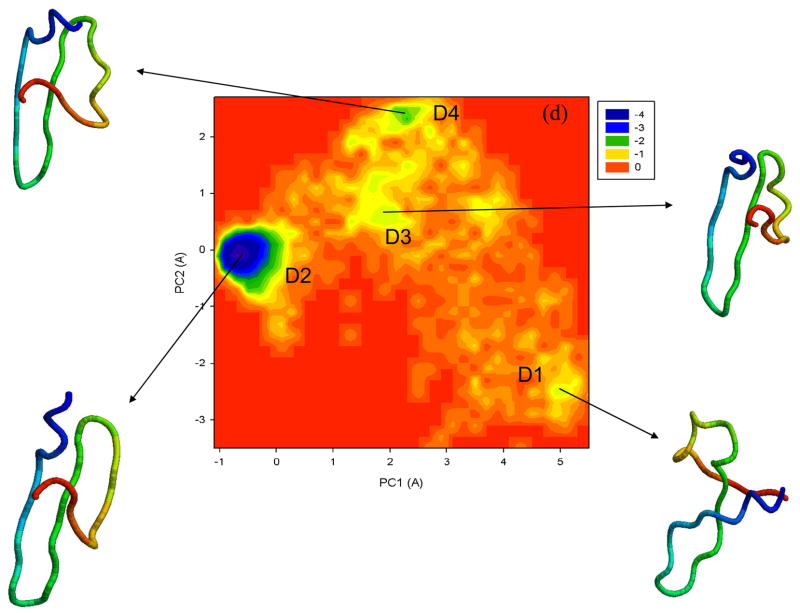
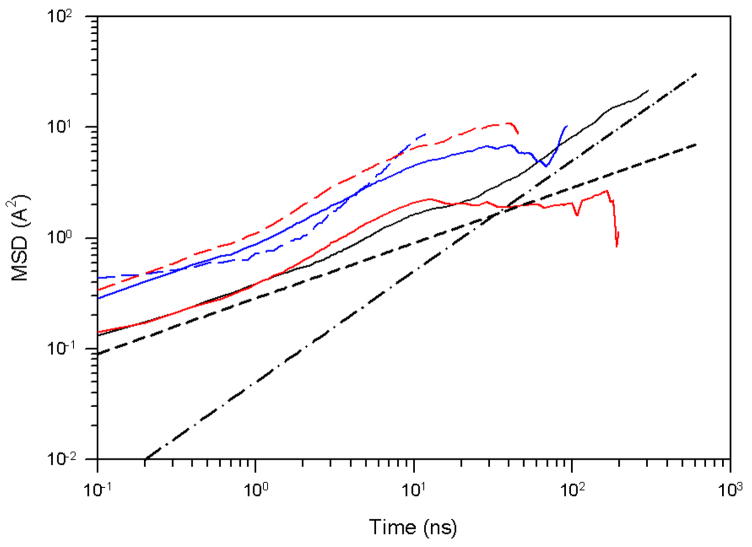
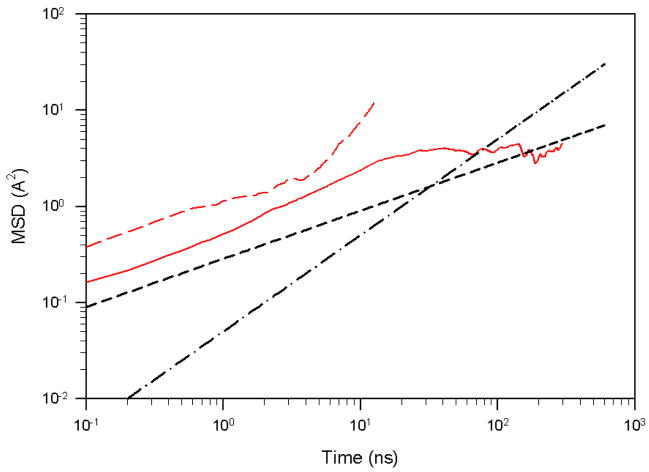
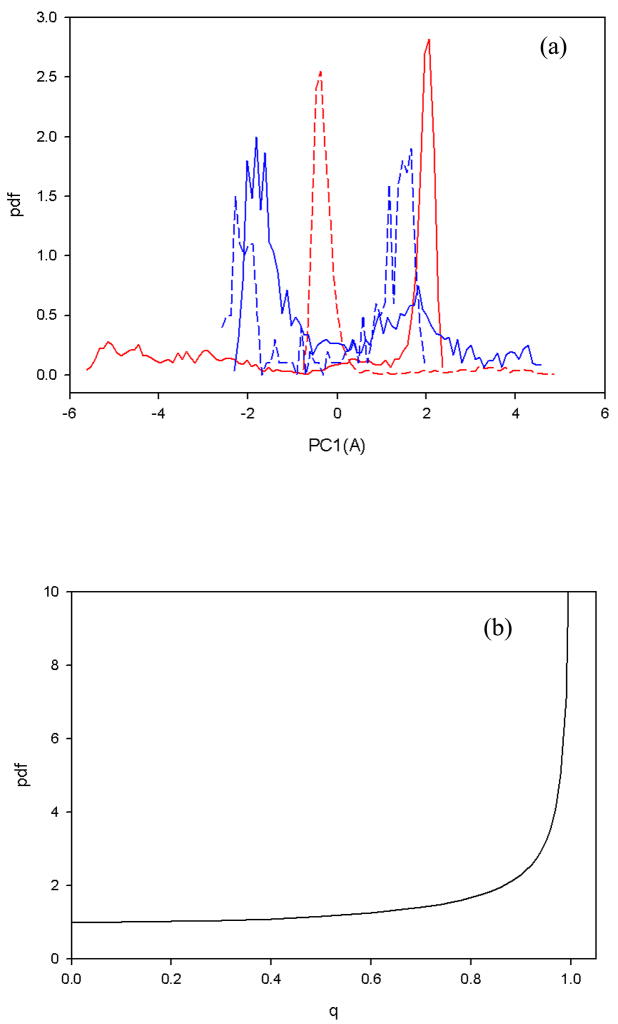
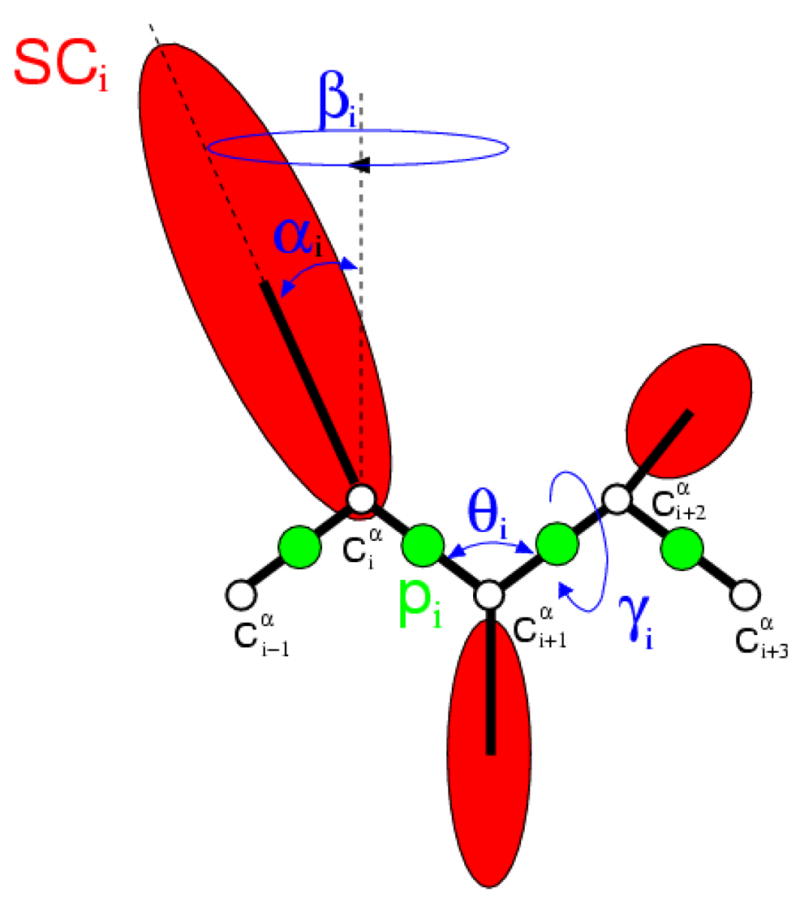
Protein folding is considered here by studying the dynamics of the folding of the triple beta-strand WW domain from the Formin-binding protein 28. Starting from the unfolded state and ending either in the native or nonnative conformational states, trajectories are generated with the coarse-grained united residue (UNRES) force field. The effectiveness of principal components analysis (PCA), an already established mathematical technique for finding global, correlated motions in atomic simulations of proteins, is evaluated here for coarse-grained trajectories. The problems related to PCA and their solutions are discussed. The folding and nonfolding of proteins are examined with free-energy landscapes. Detailed analyses of many folding and nonfolding trajectories at different temperatures show that PCA is very efficient for characterizing the general folding and nonfolding features of proteins. It is shown that the first principal component captures and describes in detail the dynamics of a system. Anomalous diffusion in the folding/nonfolding dynamics is examined by the mean-square displacement (MSD) and the fractional diffusion and fractional kinetic equations. The collisionless (or ballistic) behavior of a polypeptide undergoing Brownian motion along the first few principal components is accounted for.
Figures


References
-
- Macias MJ, Gervais V, Civera C, Oschkinat H. Structural analysis of WW domains and design of a WW prototype. Nat Struct Biol. 2000;7:375–379. - PubMed
-
- Serpell LC. Alzheimer’s amyloid fibrils: structure and assembly. Biochim Biophys Acta. 2000;1502:16–30. - PubMed
-
- Sudol M. The WW domain binds polyprolines and is involved in human diseases. Exp Mol Med. 1996;28:65–69.
-
- Passani LA, Bedford MT, Faber PW, McGinnis KM, Sharp AH, Gusella JF, Vonsattel JP, MacDonald ME. Huntingtin’s WW domain partners in Huntington’s disease post-mortem brain fulfill genetic criteria for direct involvement in Huntington’s disease pathogenesis. Human Mol Gen. 2000;9:2175–2182. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
