

Disruption of the NHR4 domain structure in AML1-ETO abrogates SON binding and promotes leukemogenesis
- PMID: 18952841
- PMCID: PMC2579385
- DOI: 10.1073/pnas.0802696105
Disruption of the NHR4 domain structure in AML1-ETO abrogates SON binding and promotes leukemogenesis
Abstract
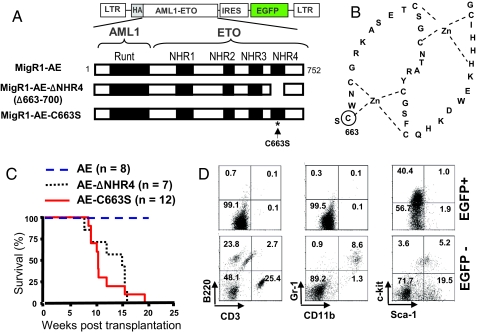
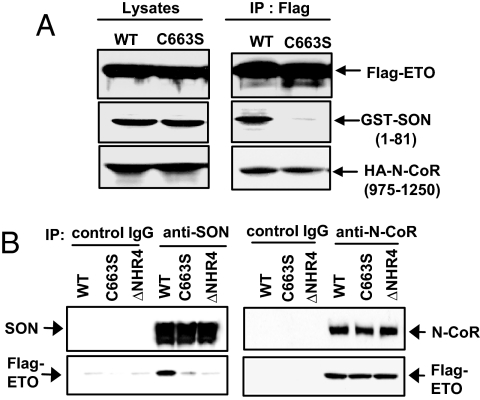
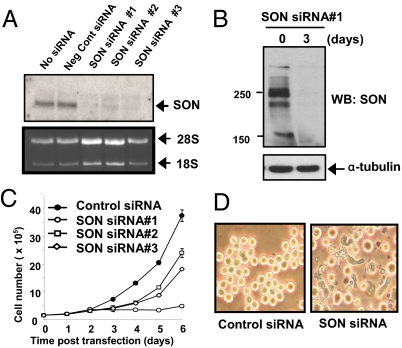
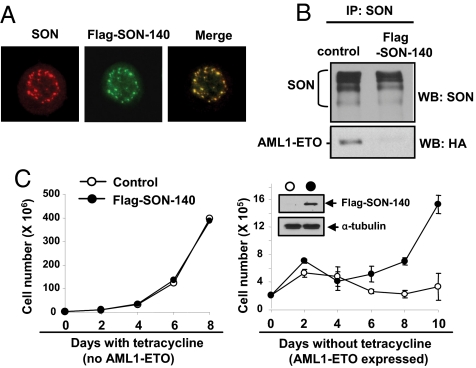
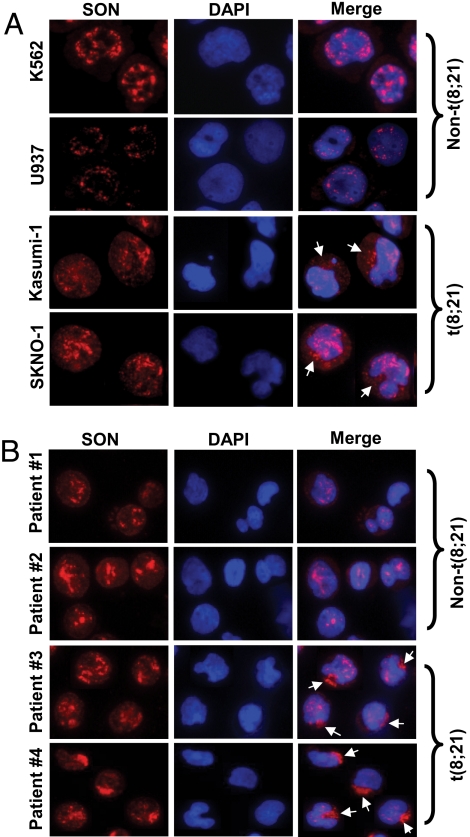
AML1-ETO is generated from t(8;21)(q22;q22), which is a common form of chromosomal translocation associated with development of acute myeloid leukemia (AML). Although full-length AML1-ETO alone fails to promote leukemia because of its detrimental effects on cell proliferation, an alternatively spliced isoform, AML1-ETO9a, without its C-terminal NHR3/NHR4 domains, strongly induces leukemia. However, full-length AML1-ETO is a major form of fusion product in many t(8;21) AML patients, suggesting additional molecular mechanisms of t(8;21)-related leukemogenesis. Here, we report that disruption of the zinc-chelating structure in the NHR4 domain of AML1-ETO by replacing only one critical amino acid leads to rapid onset of leukemia, demonstrating that the NHR4 domain with the intact structure generates inhibitory effects on leukemogenesis. Furthermore, we identified SON, a DNA/RNA-binding domain containing protein, as a novel NHR4-interacting protein. Knock-down of SON by siRNA resulted in significant growth arrest, and disruption of the interaction between AML1-ETO and endogenous SON rescued cells from AML1-ETO-induced growth arrest, suggesting that SON is an indispensable factor for cell growth, and AML1-ETO binding to SON may trigger signals inhibiting leukemogenesis. In t(8;21) AML patient-derived primary leukemic cells and cell lines, abnormal cytoplasmic localization of SON was detected, which may keep cells proliferating in the presence of full-length AML1-ETO. These results uncovered the crucial role of the NHR4 domain in determination of cellular fate during AML1-ETO-associated leukemogenesis.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Rowley JD. Chromosome translocations: Dangerous liaisons revisited. Nat Rev Cancer. 2001;1:245–250. - PubMed
-
- Langabeer SE, et al. Incidence of AML1/ETO fusion transcripts in patients entered into the MRC AML trials. MRC Adult Leukaemia Working Party. Br J Haematol. 1997;99:925–928. - PubMed
-
- Nucifora G, Rowley JD. AML1 and the 8;21 and 3;21 translocations in acute and chronic myeloid leukemia. Blood. 1995;86:1–14. - PubMed
-
- Rege K, et al. Disease features in acute myeloid leukemia with t(8;21)(q22;q22). Influence of age, secondary karyotype abnormalities, CD19 status, and extramedullary leukemia on survival. Leuk Lymphoma. 2000;40:67–77. - PubMed
-
- Rowe D, et al. Cytogenetically cryptic AML1-ETO and CBF beta-MYH11 gene rearrangements: Incidence in 412 cases of acute myeloid leukaemia. Br J Haematol. 2000;111:1051–1056. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
