

RAId_DbS: mass-spectrometry based peptide identification web server with knowledge integration
- PMID: 18954448
- PMCID: PMC2605478
- DOI: 10.1186/1471-2164-9-505
RAId_DbS: mass-spectrometry based peptide identification web server with knowledge integration
Abstract
Background: Existing scientific literature is a rich source of biological information such as disease markers. Integration of this information with data analysis may help researchers to identify possible controversies and to form useful hypotheses for further validations. In the context of proteomics studies, individualized proteomics era may be approached through consideration of amino acid substitutions/modifications as well as information from disease studies. Integration of such information with peptide searches facilitates speedy, dynamic information retrieval that may significantly benefit clinical laboratory studies.
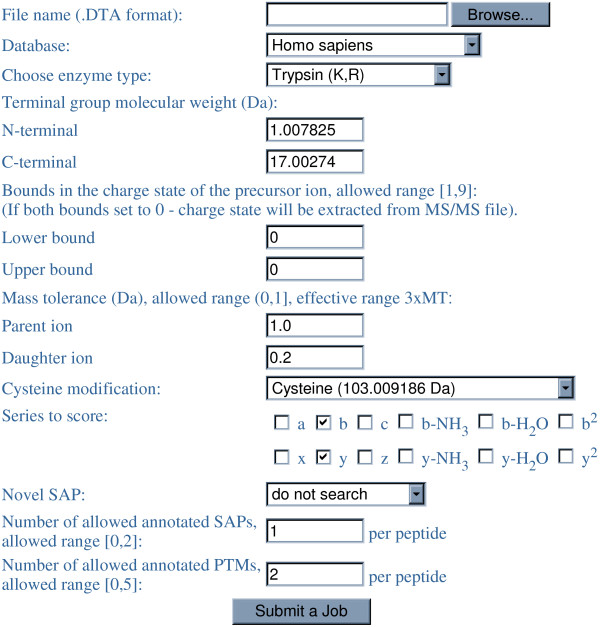
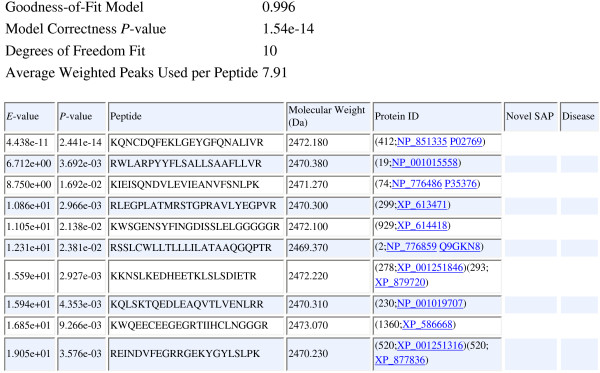
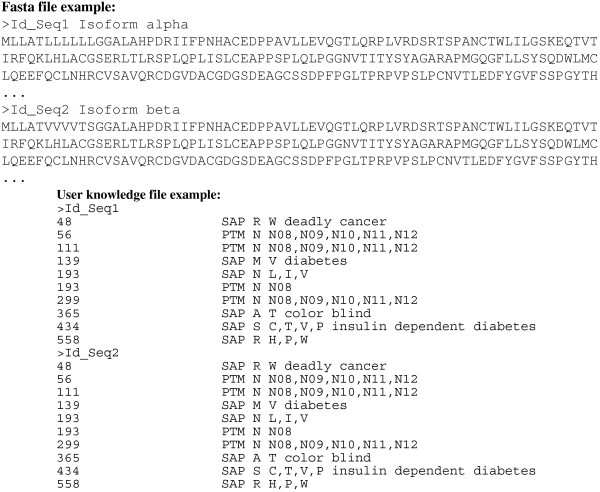
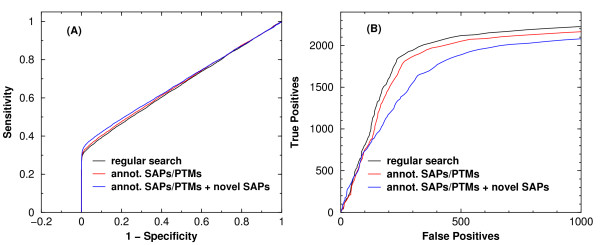
Description: We have integrated from various sources annotated single amino acid polymorphisms, post-translational modifications, and their documented disease associations (if they exist) into one enhanced database per organism. We have also augmented our peptide identification software RAId_DbS to take into account this information while analyzing a tandem mass spectrum. In principle, one may choose to respect or ignore the correlation of amino acid polymorphisms/modifications within each protein. The former leads to targeted searches and avoids scoring of unnecessary polymorphism/modification combinations; the latter explores possible polymorphisms in a controlled fashion. To facilitate new discoveries, RAId_DbS also allows users to conduct searches permitting novel polymorphisms as well as to search a knowledge database created by the users.
Conclusion: We have finished constructing enhanced databases for 17 organisms. The web link to RAId_DbS and the enhanced databases is http://www.ncbi.nlm.nih.gov/CBBResearch/qmbp/RAId_DbS/index.html. The relevant databases and binaries of RAId_DbS for Linux, Windows, and Mac OS X are available for download from the same web page.
Figures



References
-
- Collins FS, Brooks LD, Chakravarti A. A DNA polymorphism discovery resource for research on human genetic variation. Genome Res. 1998;8:1229–1231. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
