

Protein functional surfaces: global shape matching and local spatial alignments of ligand binding sites
- PMID: 18954462
- PMCID: PMC2626596
- DOI: 10.1186/1472-6807-8-45
Protein functional surfaces: global shape matching and local spatial alignments of ligand binding sites
Abstract
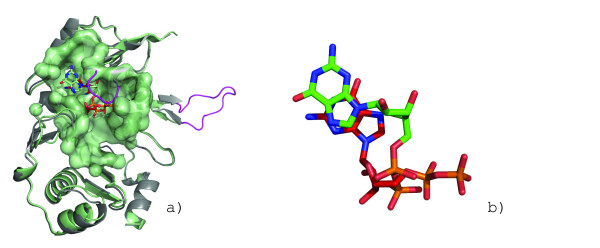
Background: Protein surfaces comprise only a fraction of the total residues but are the most conserved functional features of proteins. Surfaces performing identical functions are found in proteins absent of any sequence or fold similarity. While biochemical activity can be attributed to a few key residues, the broader surrounding environment plays an equally important role.
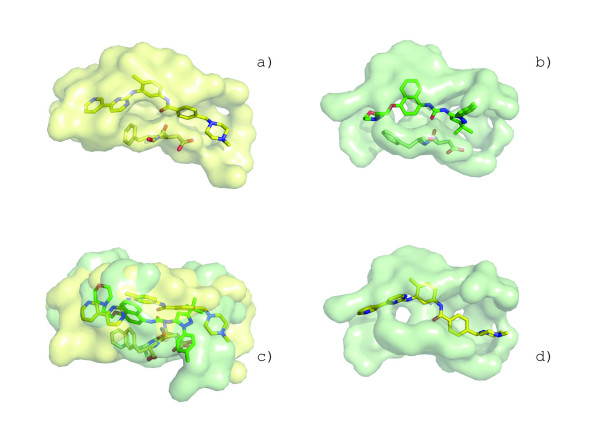
Results: We describe a methodology that attempts to optimize two components, global shape and local physicochemical texture, for evaluating the similarity between a pair of surfaces. Surface shape similarity is assessed using a three-dimensional object recognition algorithm and physicochemical texture similarity is assessed through a spatial alignment of conserved residues between the surfaces. The comparisons are used in tandem to efficiently search the Global Protein Surface Survey (GPSS), a library of annotated surfaces derived from structures in the PDB, for studying evolutionary relationships and uncovering novel similarities between proteins.
Conclusion: We provide an assessment of our method using library retrieval experiments for identifying functionally homologous surfaces binding different ligands, functionally diverse surfaces binding the same ligand, and binding surfaces of ubiquitous and conformationally flexible ligands. Results using surface similarity to predict function for proteins of unknown function are reported. Additionally, an automated analysis of the ATP binding surface landscape is presented to provide insight into the correlation between surface similarity and function for structures in the PDB and for the subset of protein kinases.
Figures













References
-
- Ye Y, Li Z, Godzik A. Modeling and analyzing three-dimensional structures of human disease proteins. Pac Symp Biocomput. 2006:439–450. - PubMed
-
- Artymiuk PJ, Poirrette AR, Grindley HM, Rice DW, Willett P. A graph-theoretic approach to the identification of three-dimensional patterns of amino acid side-chains in protein structures. J Mol Biol. 1994;243:327–344. - PubMed
-
- Schmitt S, Kuhn D, Klebe G. A new method to detect related function among proteins independent of sequence and fold homology. J Mol Biol. 2002;323:387–406. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
