

Structure of the yeast vacuolar ATPase
- PMID: 18955482
- PMCID: PMC2602884
- DOI: 10.1074/jbc.M805345200
Structure of the yeast vacuolar ATPase
Abstract
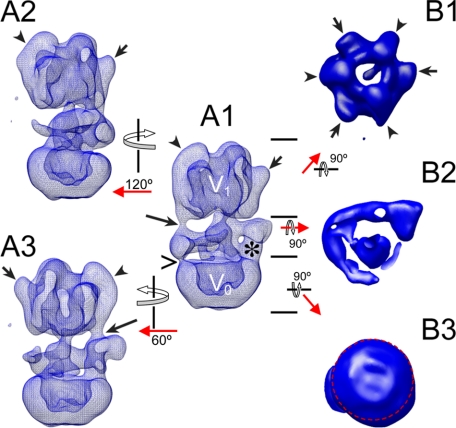
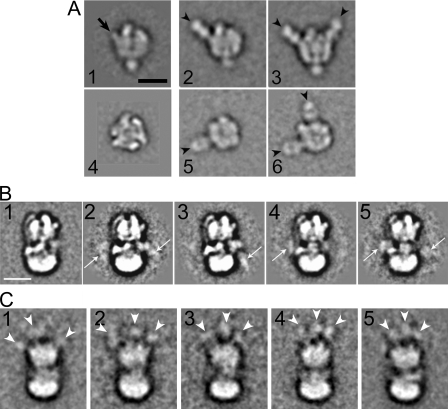
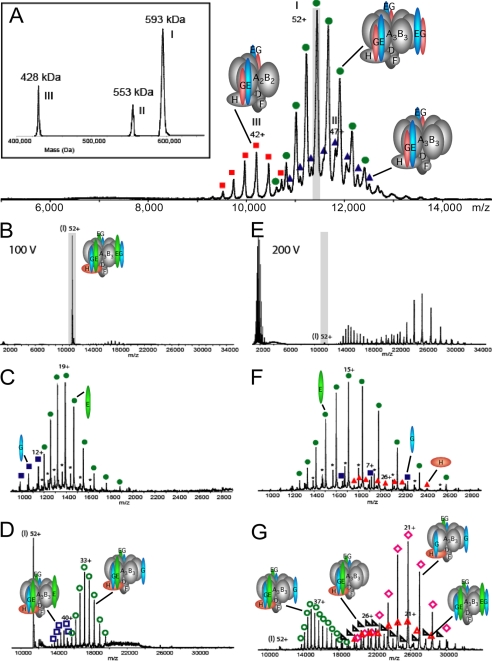
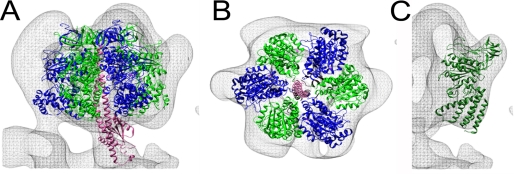
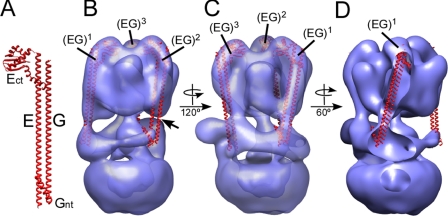
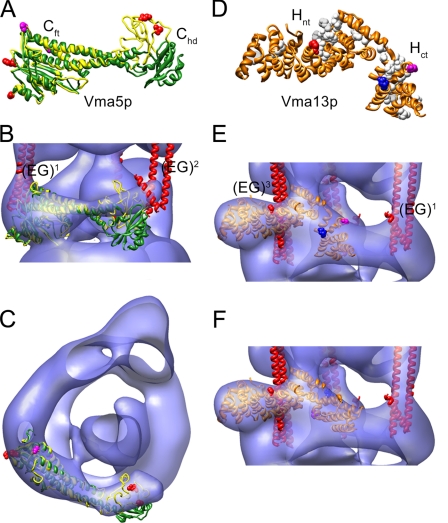
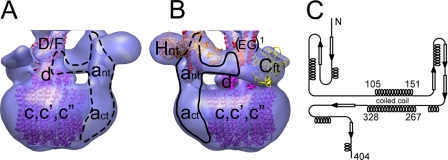
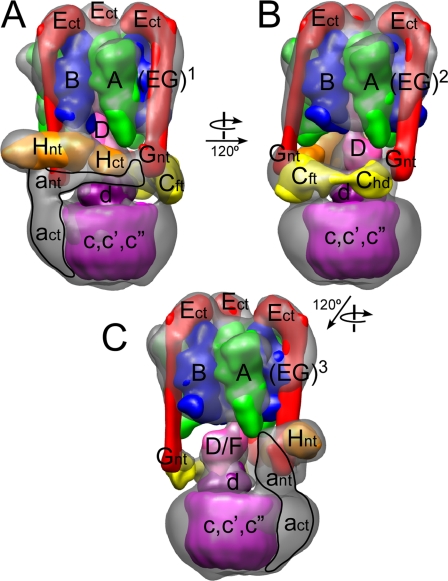
The subunit architecture of the yeast vacuolar ATPase (V-ATPase) was analyzed by single particle transmission electron microscopy and electrospray ionization (ESI) tandem mass spectrometry. A three-dimensional model of the intact V-ATPase was calculated from two-dimensional projections of the complex at a resolution of 25 angstroms. Images of yeast V-ATPase decorated with monoclonal antibodies against subunits A, E, and G position subunit A within the pseudo-hexagonal arrangement in the V1, the N terminus of subunit G in the V1-V0 interface, and the C terminus of subunit E at the top of the V1 domain. ESI tandem mass spectrometry of yeast V1-ATPase showed that subunits E and G are most easily lost in collision-induced dissociation, consistent with a peripheral location of the subunits. An atomic model of the yeast V-ATPase was generated by fitting of the available x-ray crystal structures into the electron microscopy-derived electron density map. The resulting atomic model of the yeast vacuolar ATPase serves as a framework to help understand the role the peripheral stalk subunits are playing in the regulation of the ATP hydrolysis driven proton pumping activity of the vacuolar ATPase.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
