

Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death
- PMID: 18955568
- PMCID: PMC2585850
- DOI: 10.1084/jem.20080767
Lipid mediators in innate immunity against tuberculosis: opposing roles of PGE2 and LXA4 in the induction of macrophage death
Abstract
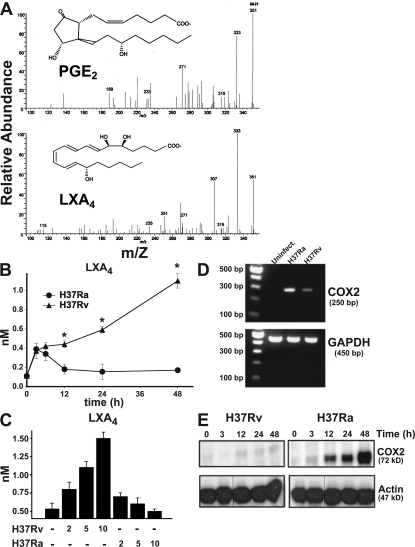
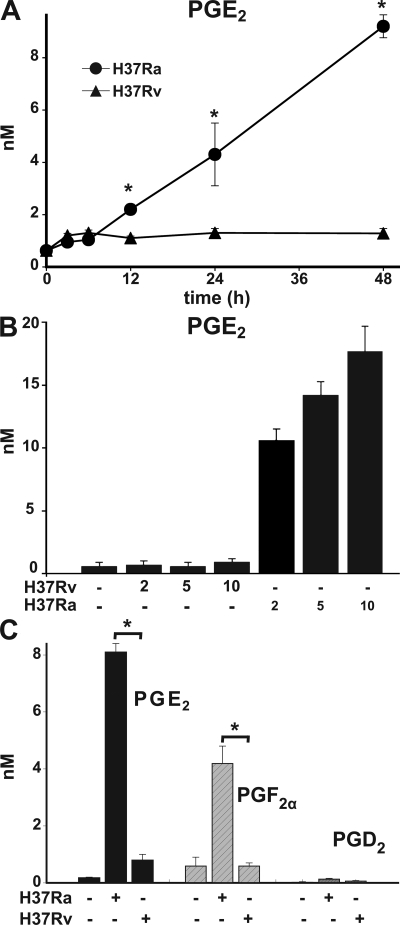
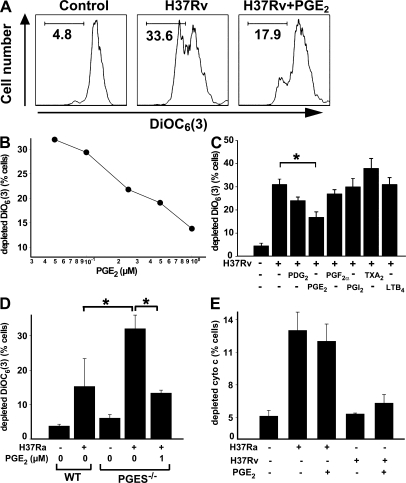
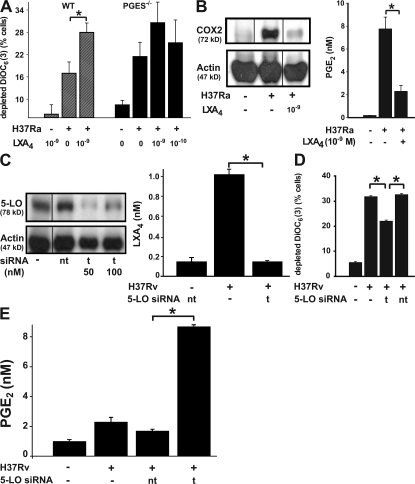
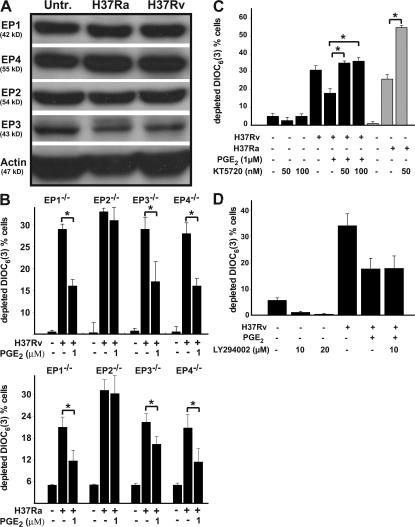
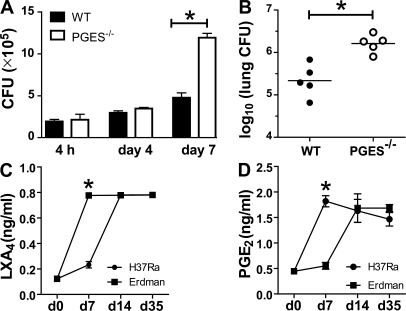
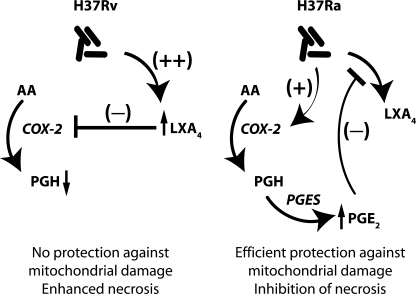
Virulent Mycobacterium tuberculosis (Mtb) induces a maladaptive cytolytic death modality, necrosis, which is advantageous for the pathogen. We report that necrosis of macrophages infected with the virulent Mtb strains H37Rv and Erdmann depends on predominant LXA(4) production that is part of the antiinflammatory and inflammation-resolving action induced by Mtb. Infection of macrophages with the avirulent H37Ra triggers production of high levels of the prostanoid PGE(2), which promotes protection against mitochondrial inner membrane perturbation and necrosis. In contrast to H37Ra infection, PGE(2) production is significantly reduced in H37Rv-infected macrophages. PGE(2) acts by engaging the PGE(2) receptor EP2, which induces cyclic AMP production and protein kinase A activation. To verify a role for PGE(2) in control of bacterial growth, we show that infection of prostaglandin E synthase (PGES)(-/-) macrophages in vitro with H37Rv resulted in significantly higher bacterial burden compared with wild-type macrophages. More importantly, PGES(-/-) mice harbor significantly higher Mtb lung burden 5 wk after low-dose aerosol infection with virulent Mtb. These in vitro and in vivo data indicate that PGE(2) plays a critical role in inhibition of Mtb replication.
Figures
References
-
- Corbett, E.L., C.J. Watt, N. Walker, B.G. Williams, M.C. Raviglione, and C. Dye. 2003. The growing burden of tuberculosis: global trends, and interactions with the HIV epidemic. Arch. Intern. Med. 163:1009–1021. - PubMed
-
- Dannenberg, A.M., and G.A. Rook. 1994. Pathogenesis of pulmonary tuberculosis: an interplay of tissue-damaging and macrophage-activating immune responses – dual mechanisms that control bacillary multiplication. In Tuberculosis. Pathogenesis, Protection and Control. Bloom B R, editor. American Society for Microbiology, Washington DC. 459–484.
-
- Leemans, J.C., N.P. Juffermans, S. Florquin, N. van Rooijen, M.J. Verwordeldonk, A. Verbon, S.J.H. van Deventer, T. van der Poll. 2001. Depletion of alveolar macrophages exerts protective effects in pulmonary tuberculosis in mice. J. Immunol. 166:4604–4611. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
