

Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling
- PMID: 18971204
- PMCID: PMC2638771
- DOI: 10.1093/hmg/ddn359
Twinkle mutations associated with autosomal dominant progressive external ophthalmoplegia lead to impaired helicase function and in vivo mtDNA replication stalling
Abstract
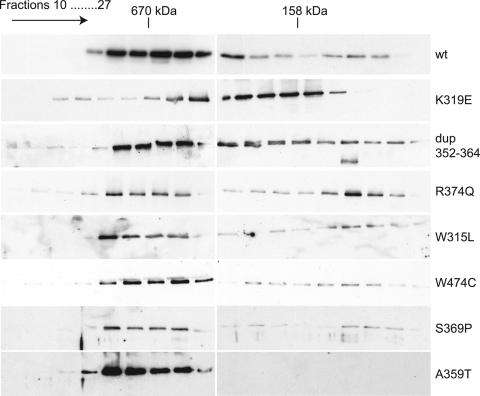
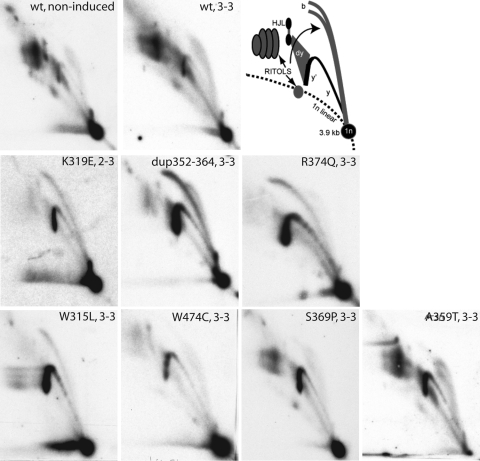
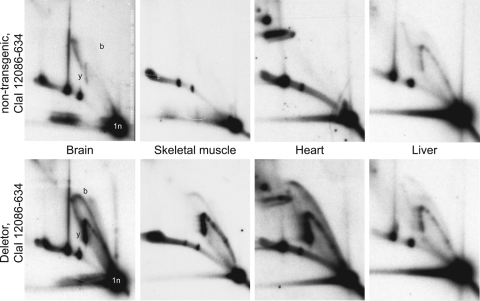
Mutations in the mitochondrial helicase Twinkle underlie autosomal dominant progressive external ophthalmoplegia (PEO), as well as recessively inherited infantile-onset spinocerebellar ataxia and rare forms of mitochondrial DNA (mtDNA) depletion syndrome. Familial PEO is typically associated with the occurrence of multiple mtDNA deletions, but the mechanism by which Twinkle dysfunction induces deletion formation has been under debate. Here we looked at the effects of Twinkle adPEO mutations in human cell culture and studied the mtDNA replication in the Deletor mouse model, which expresses a dominant PEO mutation in Twinkle and accumulates multiple mtDNA deletions during life. We show that expression of dominant Twinkle mutations results in the accumulation of mtDNA replication intermediates in cell culture. This indicated severe replication pausing or stalling and caused mtDNA depletion. A strongly enhanced accumulation of replication intermediates was evident also in six-week-old Deletor mice compared with wild-type littermates, even though mtDNA deletions accumulate in a late-onset fashion in this model. In addition, our results in cell culture pointed to a problem of transcription that preceded the mtDNA depletion phenotype and might be of relevance in adPEO pathophysiology. Finally, in vitro assays showed functional defects in the various Twinkle mutants and broadly agreed with the cell culture phenotypes such as the level of mtDNA depletion and the level of accumulation of replication intermediates. On the basis of our results we suggest that mtDNA replication pausing or stalling is the common consequence of Twinkle PEO mutations that predisposes to multiple deletion formation.
Figures
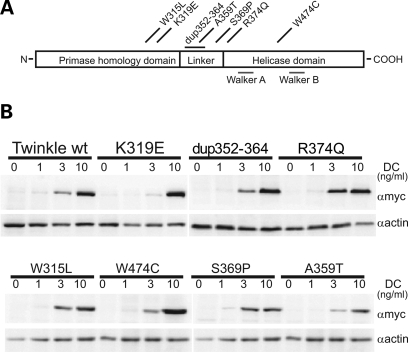
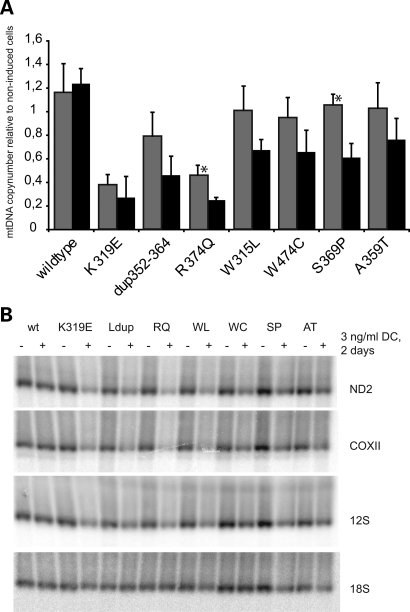
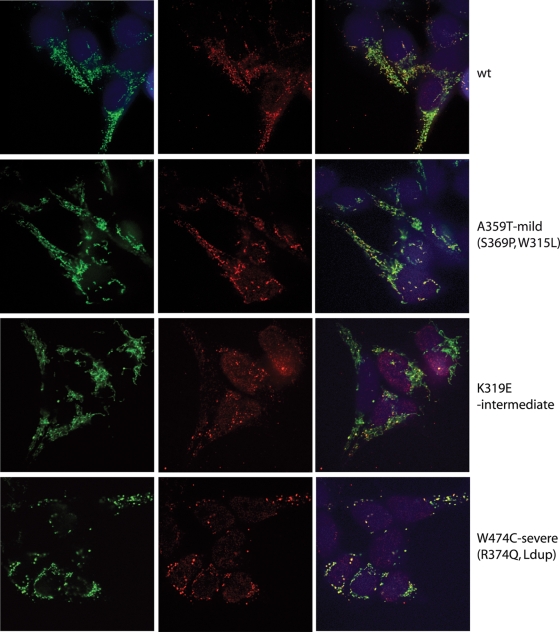
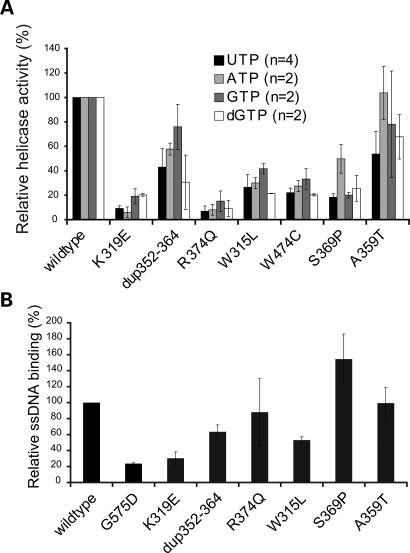
References
-
- Schaefer A.M., Blakely E.L., He L., Whittaker R.G., Taylor R.W., Chinnery P.F., Turnbull D.M. Prevalence of mitochondrial DNA disease in adults. Ann. Neurol. 2008;63:35–39. - PubMed
-
- Majamaa K., Moilanen J.S., Uimonen S., Remes A.M., Salmela P.I., Karppa M., Majamaa-Voltti K.A., Rusanen H., Sorri M., Peuhkurinen K.J., et al. Epidemiology of A3243G, the mutation for mitochondrial encephalomyopathy, lactic acidosis, and strokelike episodes: prevalence of the mutation in an adult population. Am. J. Hum. Genet. 1998;63:447–454. - PMC - PubMed
-
- Hakonen A.H., Heiskanen S., Juvonen V., Lappalainen I., Luoma P.T., Rantamaki M., Goethem G.V., Lofgren A., Hackman P., Paetau A., et al. Mitochondrial DNA polymerase W748S mutation: a common cause of autosomal recessive ataxia with ancient European origin. Am. J. Hum. Genet. 2005;77:430–441. - PMC - PubMed
-
- Winterthun S., Ferrari G., He L., Taylor R.W., Zeviani M., Turnbull D.M., Engelsen B.A., Moen G., Bindoff L.A. Autosomal recessive mitochondrial ataxic syndrome due to mitochondrial polymerase gamma mutations. Neurology. 2005;64:1204–1208. - PubMed
-
- Naviaux R.K., Nguyen K.V. POLG mutations associated with Alpers’ syndrome and mitochondrial DNA depletion. Ann. Neurol. 2004;55:706–712. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
