

Host cell factors and functions involved in vesicular stomatitis virus entry
- PMID: 18971266
- PMCID: PMC2612308
- DOI: 10.1128/JVI.01864-08
Host cell factors and functions involved in vesicular stomatitis virus entry
Abstract
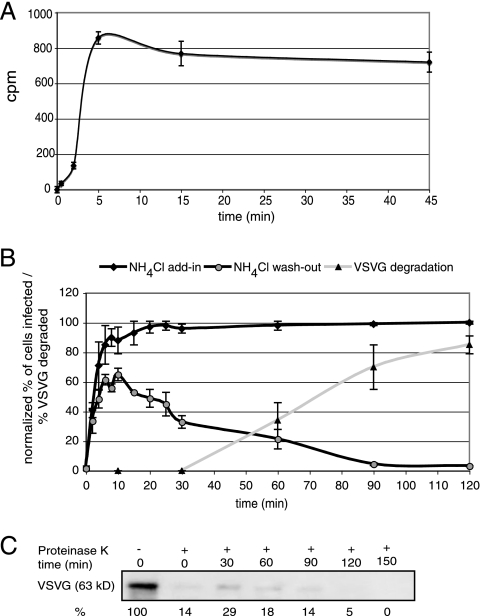
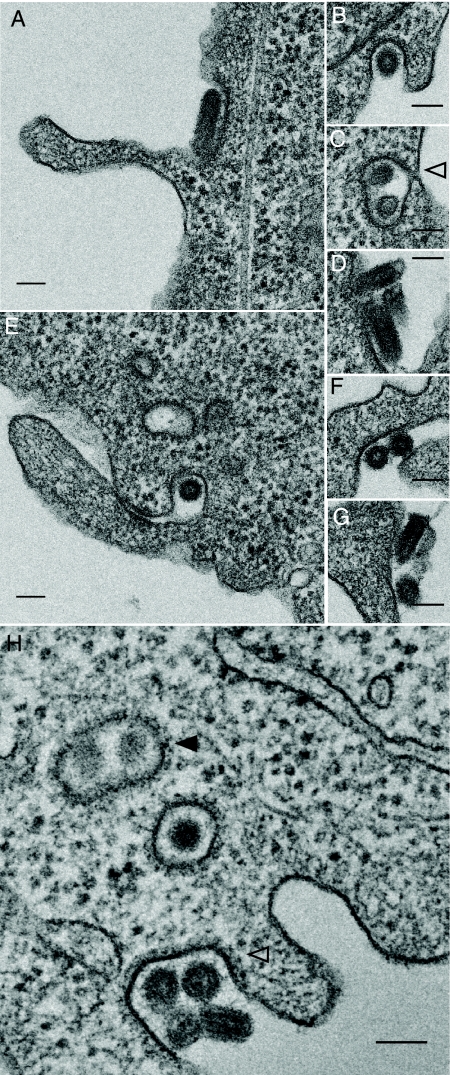
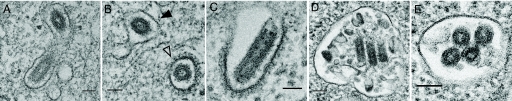
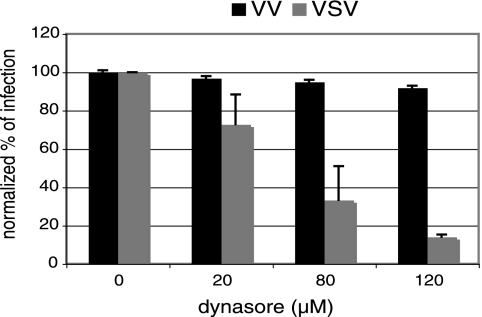
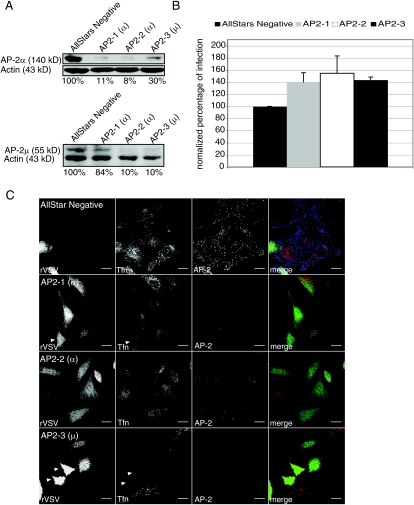
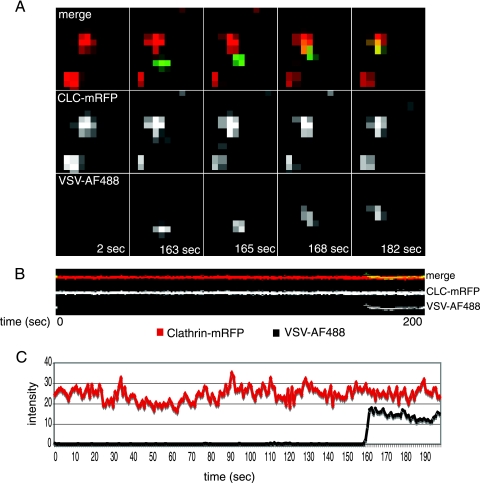
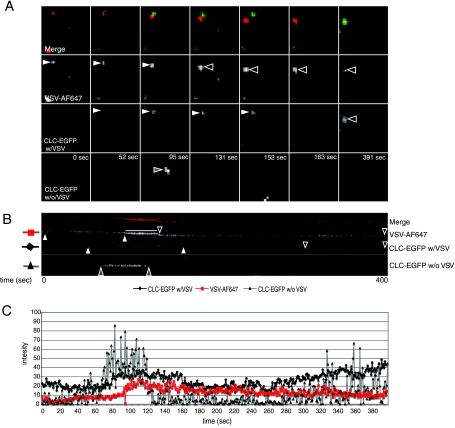
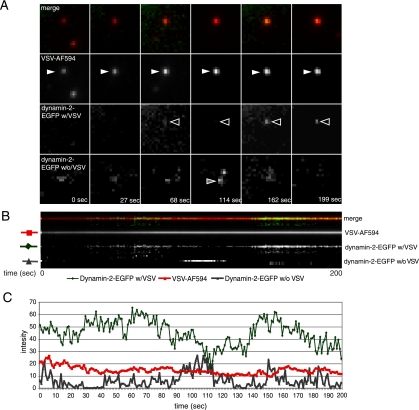
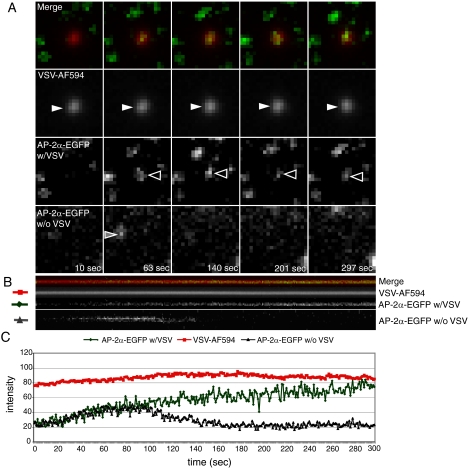
Vesicular stomatitis virus (VSV) is an animal virus that based on electron microscopy and its dependence on acidic cellular compartments for infection is thought to enter its host cells in a clathrin-dependent manner. The exact cellular mechanism, however, is largely unknown. In this study, we characterized the entry kinetics of VSV and elucidated viral requirements for host cell factors during infection in HeLa cells. We found that endocytosis of VSV was a fast process with a half time of 2.5 to 3 min and that acid activation occurred within 1 to 2 min after internalization in early endosomes. The majority of viral particles were endocytosed in a clathrin-based, dynamin-2-dependent manner. Although associated with some of the surface-bound viruses, the classical adaptor protein complex AP-2 was not required for infection. Time-lapse microscopy revealed that the virus either entered preformed clathrin-coated pits or induced de novo formation of pits. Dynamin-2 was recruited to plasma membrane-confined virus particles. Thus, VSV can induce productive internalization by exploiting a specific combination of the clathrin-associated proteins and cellular functions.
Figures
References
-
- Burns, J. C., T. Friedmann, W. Driever, M. Burrascano, and J. K. Yee. 1993. Vesicular stomatitis virus G glycoprotein pseudotyped retroviral vectors: concentration to very high titer and efficient gene transfer into mammalian and nonmammalian cells. Proc. Natl. Acad. Sci. USA 908033-8037. - PMC - PubMed
-
- Carneiro, F. A., A. S. Ferradosa, and A. T. Da Poian. 2001. Low pH-induced conformational changes in vesicular stomatitis virus glycoprotein involve dramatic structure reorganization. J. Biol. Chem. 27662-67. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
