

DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression
- PMID: 18971312
- PMCID: PMC2593572
- DOI: 10.1101/gr.074070.107
DNA methylation profile of tissue-dependent and differentially methylated regions (T-DMRs) in mouse promoter regions demonstrating tissue-specific gene expression
Abstract
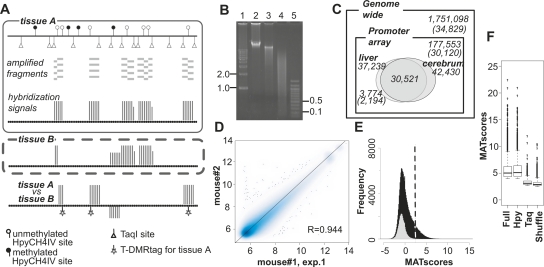
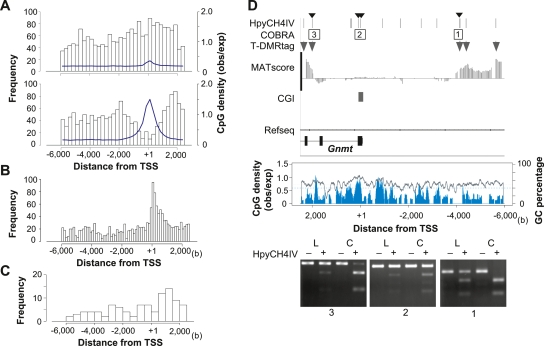
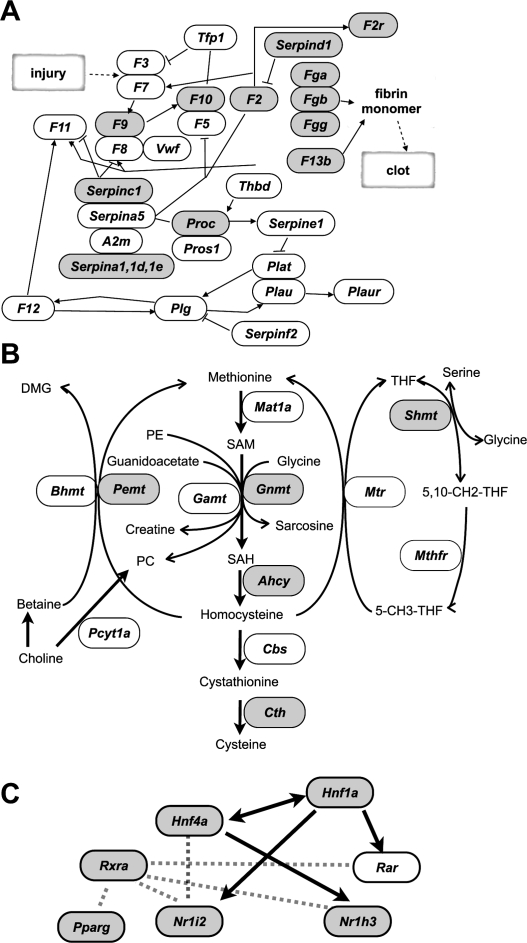
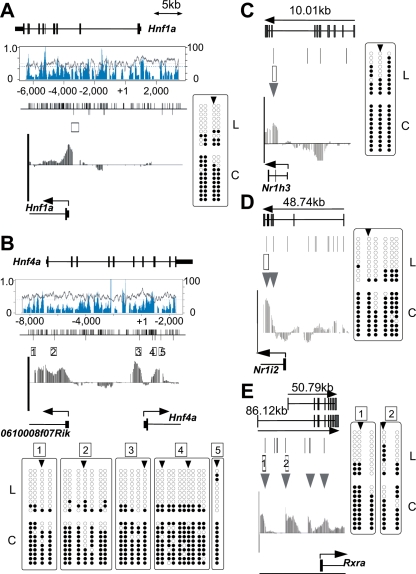
DNA methylation constitutes an important epigenetic regulation mechanism in many eukaryotes, although the extent of DNA methylation in the regulation of gene expression in the mammalian genome is poorly understood. We developed D-REAM, a genome-wide DNA methylation analysis method for tissue-dependent and differentially methylated region (T-DMR) profiling with restriction tag-mediated amplification in mouse tissues and cells. Using a mouse promoter tiling array covering a region from -6 to 2.5 kb ( approximately 30,000 transcription start sites), we found that over 3000 T-DMRs are hypomethylated in liver compared to cerebrum. The DNA methylation profile of liver was distinct from that of kidney and spleen. This hypomethylation profile marked genes that are specifically expressed in liver, including key transcription factors such as Hnf1a and Hnf4a. Genes with T-DMRs, especially those lacking CpG islands and those with HNF-1A binding motifis in their promoters, showed good correlation between their tissue-specific expression and liver hypomethylation status. T-DMRs located downstream from their transcription start sites also showed tissue-specific gene expression. These data indicate that multilayered regulation of tissue-specific gene function could be elucidated by DNA methylation tissue profiling.
Figures
References
-
- Barski A., Cuddapah S., Cui K., Roh T., Schones D., Wang Z., Wei G., Chepelev I., Zhao K. High-resolution profiling of histone methylations in the human genome. Cell. 2007;129:823–837. - PubMed
-
- Bird A., Taggart M., Frommer M., Miller O., Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell. 1985;40:91–99. - PubMed
-
- Bock C., Reither S., Mikeska T., Paulsen M., Walter J., Lengauer T. BiQ Analyzer: Visualization and quality control for DNA methylation data from bisulfite sequencing. Bioinformatics. 2005;21:4067–4068. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases