

Requirement of 3-phosphoinositide-dependent protein kinase-1 for BDNF-mediated neuronal survival
- PMID: 18971483
- PMCID: PMC3383049
- DOI: 10.1523/JNEUROSCI.2135-08.2008
Requirement of 3-phosphoinositide-dependent protein kinase-1 for BDNF-mediated neuronal survival
Abstract
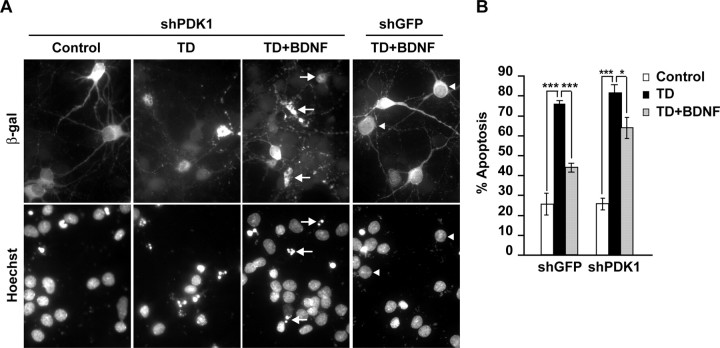
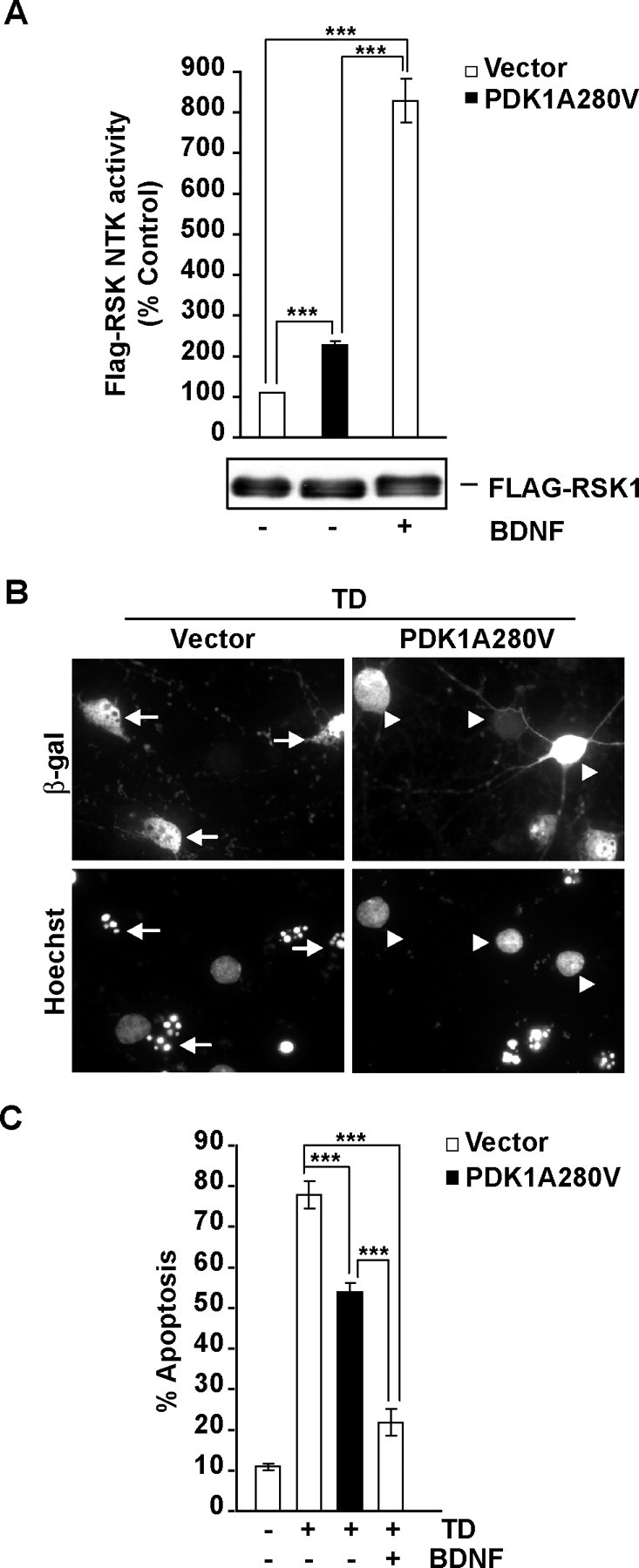
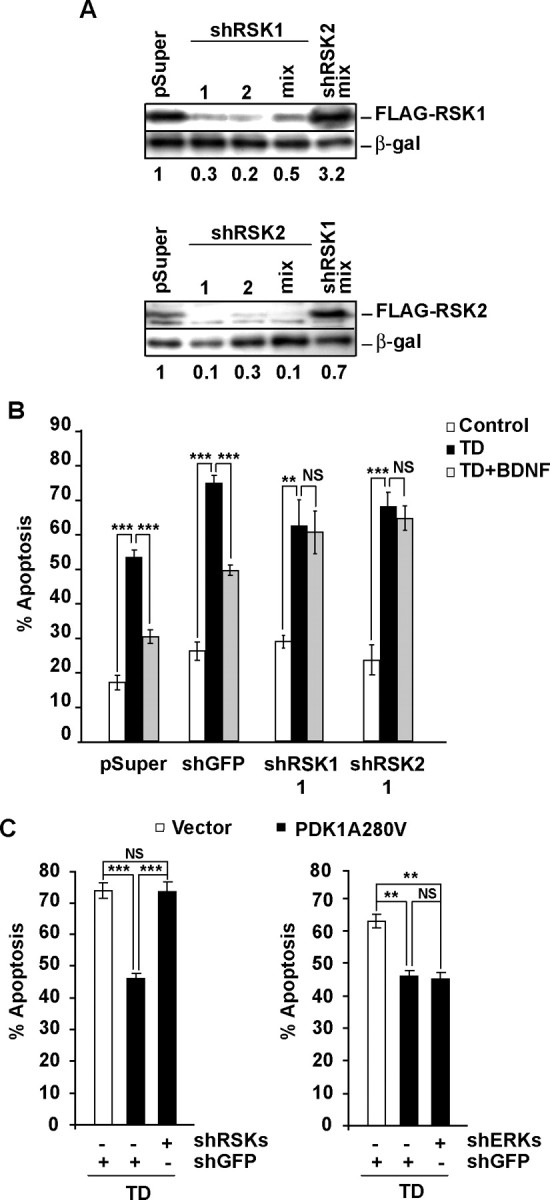
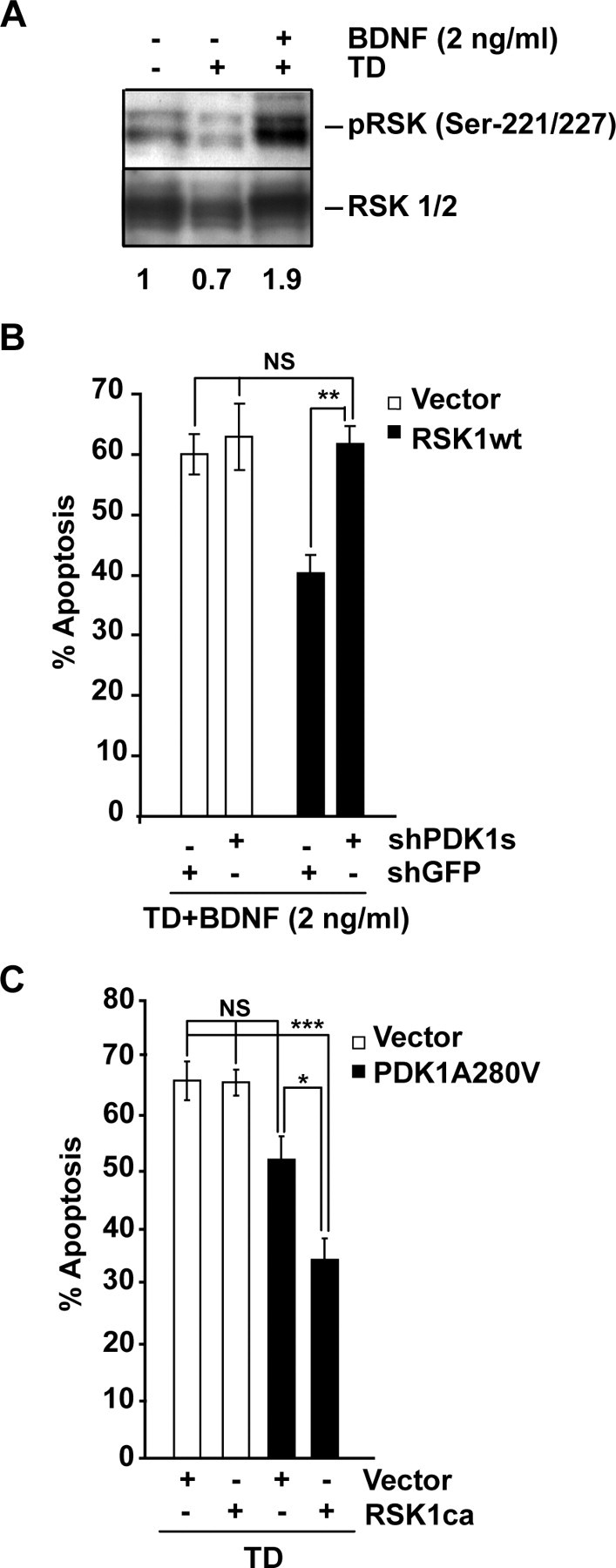
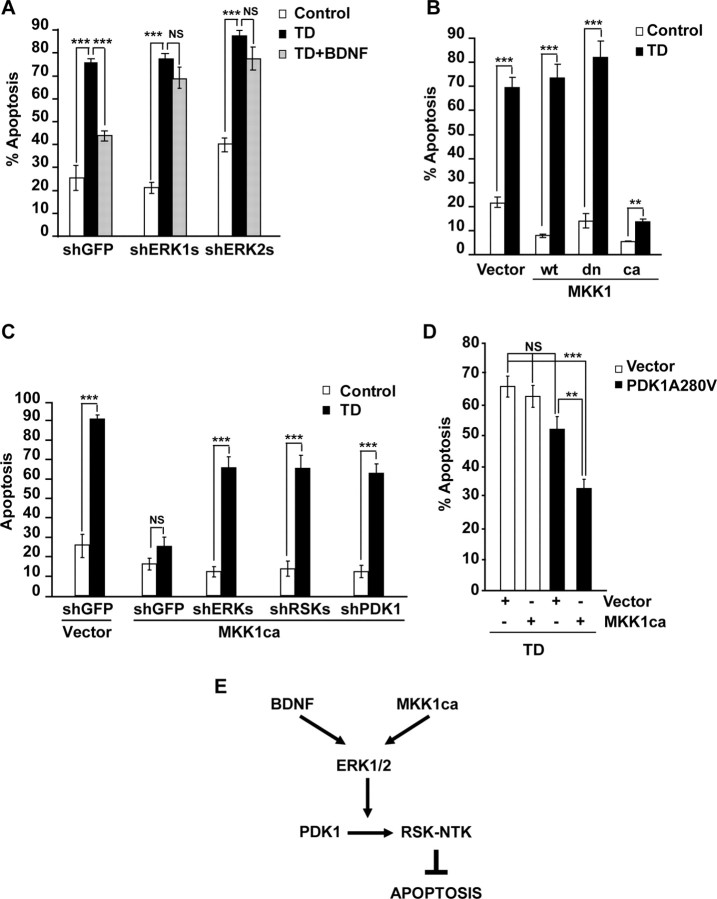
Although PDK1 regulates several signaling pathways that respond to neurotrophins, direct evidence for its involvement in neurotrophin-mediated survival has not yet been reported. Here we show high neuronal expression of active PDK1 in the rat cortex and hippocampus at the developmental stages with pronounced dependence on extracellular survival signals. Also, in cultured cortical neurons from newborn rats, BDNF resulted in PDK1- and extracellular signal-regulated kinase-1/2 (ERK1/2)-mediated activation of their direct target, the p90 ribosomal S6 kinase 1/2 (RSK1/2). In trophic-deprived cortical neurons, knockdown of endogenous PDK1 attenuated the antiapoptotic survival response to 10 ng/ml BDNF, whereas an overexpressed active mutant form of PDK1 reduced apoptosis. The neuroprotection by BDNF or active PDK1 required RSK1/2. Conversely, PDK1 knockdown reversed the survival effects of combining the overexpressed RSK1 with a low, subprotective BDNF concentration of 2 ng/ml. Likewise, the protection by the overexpressed, active PDK1 was enhanced by coexpression of an active RSK1 mutant. Consistent with the observations that in BDNF-stimulated neurons RSK1/2 activation required both PDK1 and ERK1/2, ERK1/2 knockdown removed BDNF-mediated survival. Selective activation of ERK1/2 with an overexpressed active mutant form of MKK1 resulted in RSK1/2- and PDK1-dependent neuroprotection. Finally, at subprotective plasmid DNA dosage, overexpression of the active MKK1 and PDK1 mutants produced synergistic effect on survival. Our findings indicate a critical role for PDK1-RSK1/2 signaling in BDNF-mediated neuronal survival. Thus, the PDK1 is indispensable for the antiapoptotic effects of the ERK1/2 pathway offering previously unrecognized layer of survival signal processing and integration.
Figures
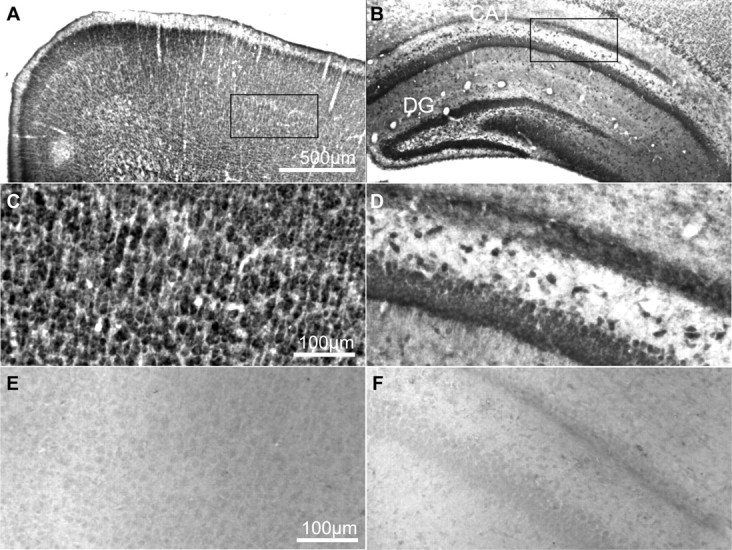
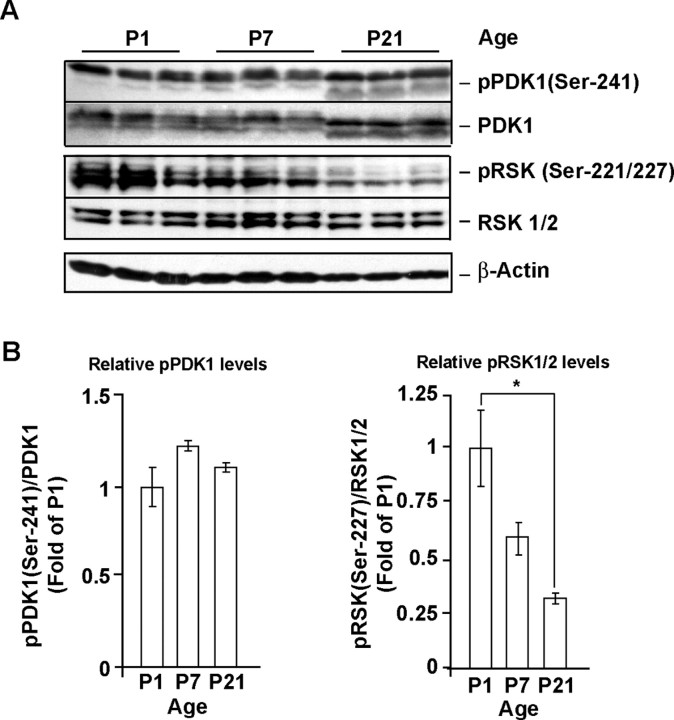
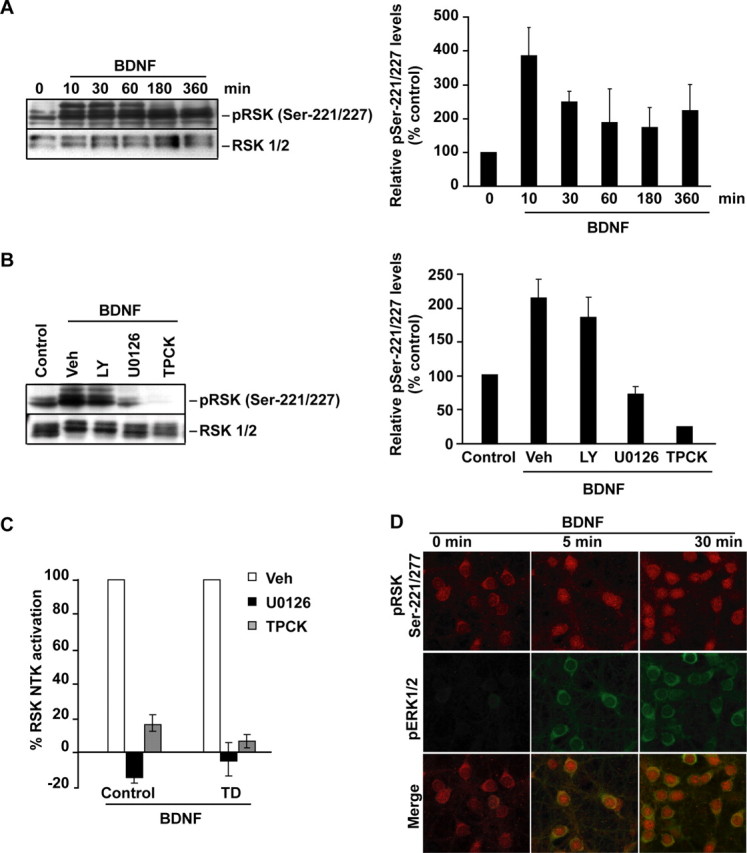
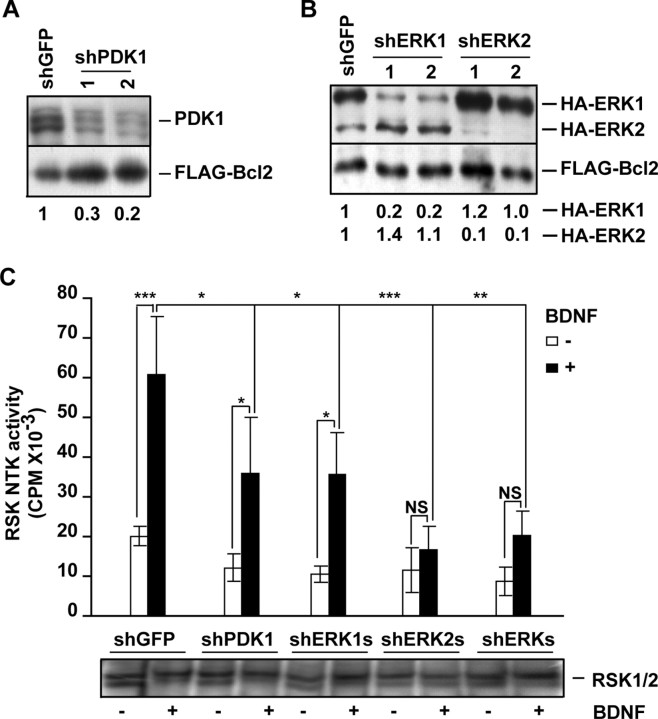
References
-
- Adams JP, Sweatt JD. Molecular psychology: roles for the ERK MAP kinase cascade in memory. Annu Rev Pharmacol Toxicol. 2002;42:135–163. - PubMed
-
- Bayascas JR, Wullschleger S, Sakamoto K, García-Martínez JM, Clacher C, Komander D, van Aalten DM, Boini KM, Lang F, Lipina C, Logie L, Sutherland C, Chudek JA, van Diepen JA, Voshol PJ, Lucocq JM, Alessi DR. Mutation of PDK1 PH domain inhibits PKB/Akt leading to small size and insulin-resistance. Mol Cell Biol. 2008;28:3258–3272. - PMC - PubMed
-
- Bonni A, Brunet A, West AE, Datta SR, Takasu MA, Greenberg ME. Cell survival promoted by the Ras-MAPK signaling pathway by transcription-dependent and -independent mechanisms. Science. 1999;286:1358–1362. - PubMed
-
- Brummelkamp TR, Bernards R, Agami R. A system for stable expression of short interfering RNAs in mammalian cells. Science. 2002;296:550–553. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous