

Calpain 3, the "gatekeeper" of proper sarcomere assembly, turnover and maintenance
- PMID: 18974005
- PMCID: PMC2614824
- DOI: 10.1016/j.nmd.2008.08.005
Calpain 3, the "gatekeeper" of proper sarcomere assembly, turnover and maintenance
Abstract
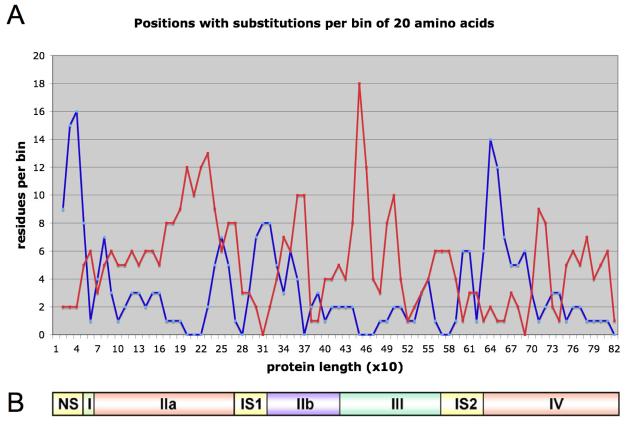
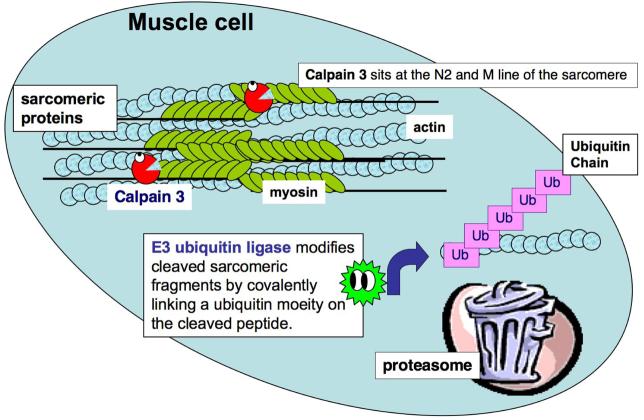
Calpain 3 is a member of the calpain family of calcium-dependent intracellular proteases. Thirteen years ago it was discovered that mutations in calpain 3 (CAPN3) result in an autosomal recessive and progressive form of limb girdle muscular dystrophy called limb girdle muscular dystrophy type 2A. While calpain 3 mRNA is expressed at high levels in muscle and appears to have some role in developmental processes, muscles of patients and mice lacking calpain 3 still form apparently normal muscle during prenatal development; thus, a functional calpain 3 protease is not mandatory for muscle to form in vivo but it is a pre-requisite for muscle to remain healthy. Despite intensive research in this field, the physiological substrates of the calpain 3 protein (hereafter referred to as CAPN3) and its alternatively spliced isoforms remain elusive. The existence of these multiple isoforms complicates the search for the physiological functions of CAPN3 and its pathophysiological role. In this review, we summarize the genetic and biochemical evidence that point to loss of function of the full-length isoform of CAPN3, also known as p94, as the pathogenic isoform. We also argue that its natural substrates must reside in its proximity within the sarcomere where it is stored in an inactive state anchored to titin. We further propose that CAPN3 has many attributes that make it ideally suited as a sensor of sarcomeric integrity and function, involved in its repair and maintenance. Loss of these CAPN3-mediated activities can explain the "progressive" development of muscular dystrophy.
Figures
References
-
- Fardeau M, Eymard B, Mignard C, Tome FM, Richard I, Beckmann JS. Chromosome 15-linked limb-girdle muscular dystrophy: clinical phenotypes in Reunion Island and French metropolitan communities. Neuromuscul Disord. 1996b;6:447–453. - PubMed
-
- Fardeau M, Hillaire D, Mignard C, et al. Juvenile limb-girdle muscular dystrophy. Clinical, histopathological and genetic data from a small community living in the Reunion Island. Brain. 1996a;119(Pt 1):295–308. - PubMed
-
- Kaplan JC, Beckmann JS, Fardeau M. Limb girdle muscular dystrophies. In: Karpati G, Hilton-Jones D, Griggs RC, editors. Disorders of Voluntary Muscles. 7th Edition Cambridge University Press; 2001. pp. 443–463.
-
- Straub V, Bushby K. The childhood limb-girdle muscular dystrophies. Semin Pediatr Neurol. 2006;13:104–114. - PubMed
-
- Erb W. About the “juvenile form” of progressive muscle atrophy and its correlation to the so-called muscle pseudohypertrophy (Ueber die “juvenile Form” der progressiven Muskelatrophie und ihre Beziehungen zur sogenannten Pseudohypertrophie der Muskeln) Deutsch Archiv Klin Med. 1884;34:467–519.
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
