

A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome
- PMID: 18976727
- PMCID: PMC2668041
- DOI: 10.1016/j.ajhg.2008.10.003
A homozygous mutation in human PRICKLE1 causes an autosomal-recessive progressive myoclonus epilepsy-ataxia syndrome
Abstract
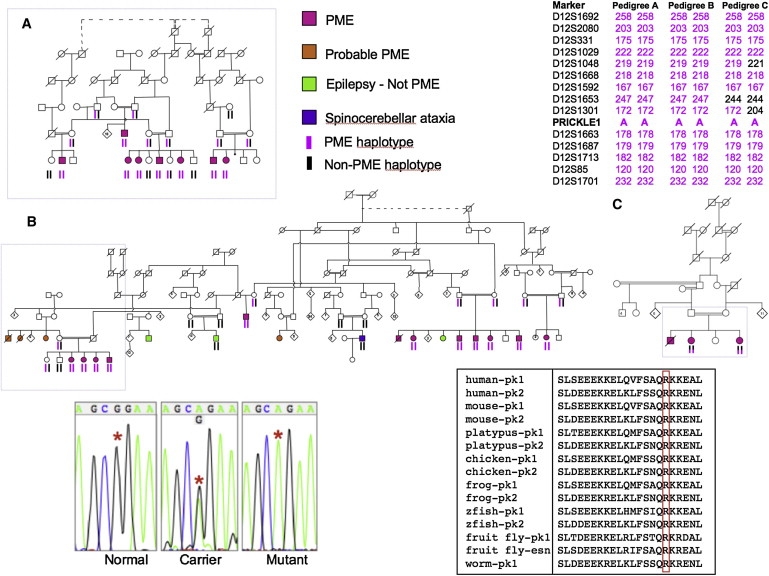
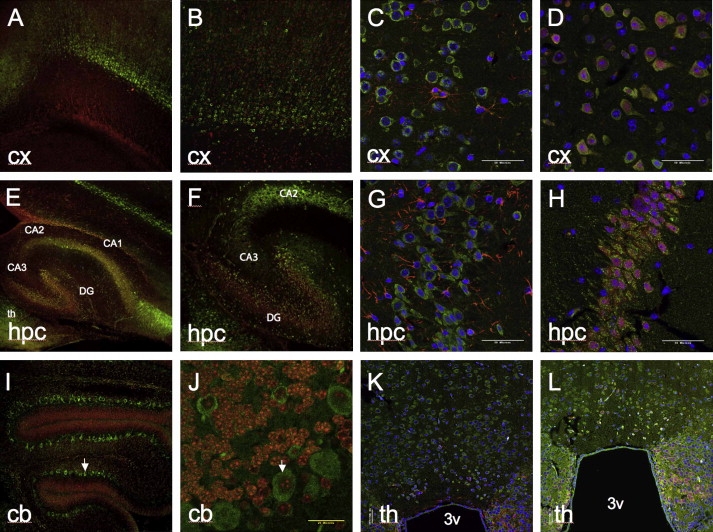
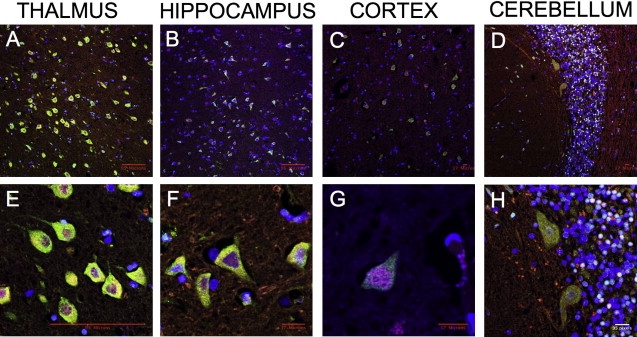
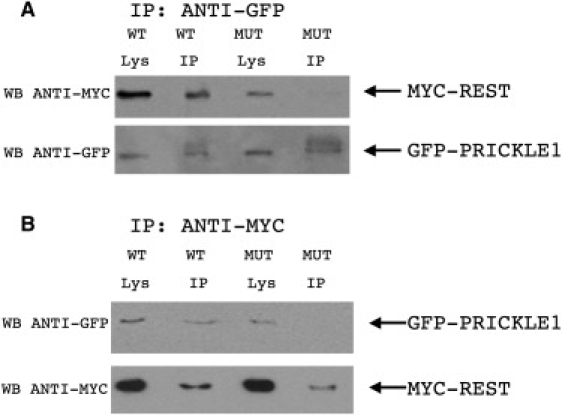
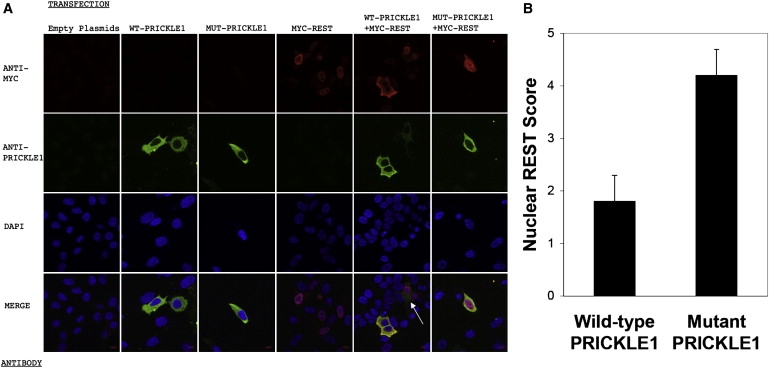
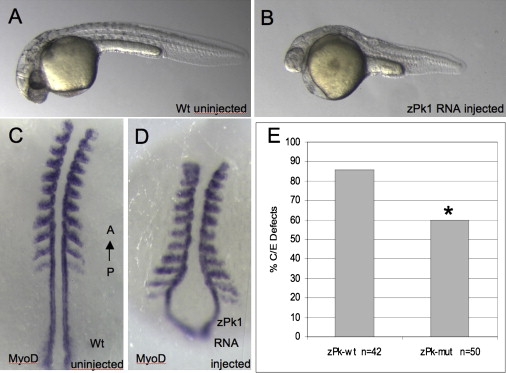
Progressive myoclonus epilepsy (PME) is a syndrome characterized by myoclonic seizures (lightning-like jerks), generalized convulsive seizures, and varying degrees of neurological decline, especially ataxia and dementia. Previously, we characterized three pedigrees of individuals with PME and ataxia, where either clinical features or linkage mapping excluded known PME loci. This report identifies a mutation in PRICKLE1 (also known as RILP for REST/NRSF interacting LIM domain protein) in all three of these pedigrees. The identified PRICKLE1 mutation blocks the PRICKLE1 and REST interaction in vitro and disrupts the normal function of PRICKLE1 in an in vivo zebrafish overexpression system. PRICKLE1 is expressed in brain regions implicated in epilepsy and ataxia in mice and humans, and, to our knowledge, is the first molecule in the noncanonical WNT signaling pathway to be directly implicated in human epilepsy.
Figures
Comment in
-
A prickly cause of progressive myoclonic epilepsy.Clin Genet. 2009 Mar;75(3):225-6. doi: 10.1111/j.1399-0004.2009.01150_1.x. Clin Genet. 2009. PMID: 19250377 No abstract available.
-
A new, progressive myoclonic epilepsy: is it a chronicle of the noncanonical or a failure to REST?Epilepsy Curr. 2009 May-Jun;9(3):82-4. doi: 10.1111/j.1535-7511.2009.01300.x. Epilepsy Curr. 2009. PMID: 19471617 Free PMC article. No abstract available.
References
-
- Berkovic S.F., Mazarib A., Walid S., Neufeld M.Y., Manelis J., Nevo Y., Korczyn A.D., Yin J., Xiong L., Pandolfo M. A new clinical and molecular form of Unverricht-Lundborg disease localized by homozygosity mapping. Brain. 2005;128:652–658. - PubMed
-
- Straussberg R., Basel-Vanagaite L., Kivity S., Dabby R., Cirak S., Nurnberg P., Voit T., Mahajnah M., Inbar D., Saifi G.M. An autosomal recessive cerebellar ataxia syndrome with upward gaze palsy, neuropathy, and seizures. Neurology. 2005;64:142–144. - PubMed
-
- Ciani L., Salinas P.C. WNTs in the vertebrate nervous system: from patterning to neuronal connectivity. Nat. Rev. Neurosci. 2005;6:351–362. - PubMed
-
- Veeman M.T., Axelrod J.D., Moon R.T. A second canon. Functions and mechanisms of beta-catenin-independent Wnt signaling. Dev. Cell. 2003;5:367–377. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
