

An integrated software system for analyzing ChIP-chip and ChIP-seq data
- PMID: 18978777
- PMCID: PMC2596672
- DOI: 10.1038/nbt.1505
An integrated software system for analyzing ChIP-chip and ChIP-seq data
Abstract
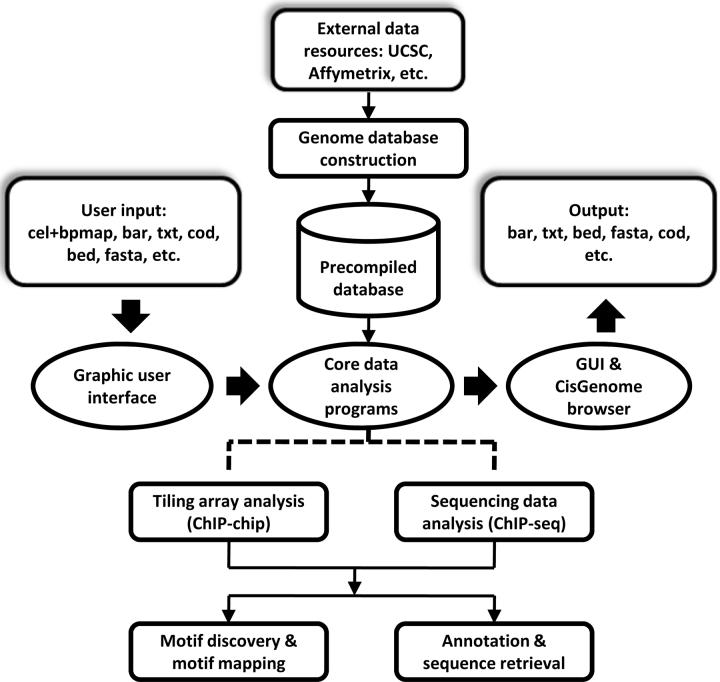
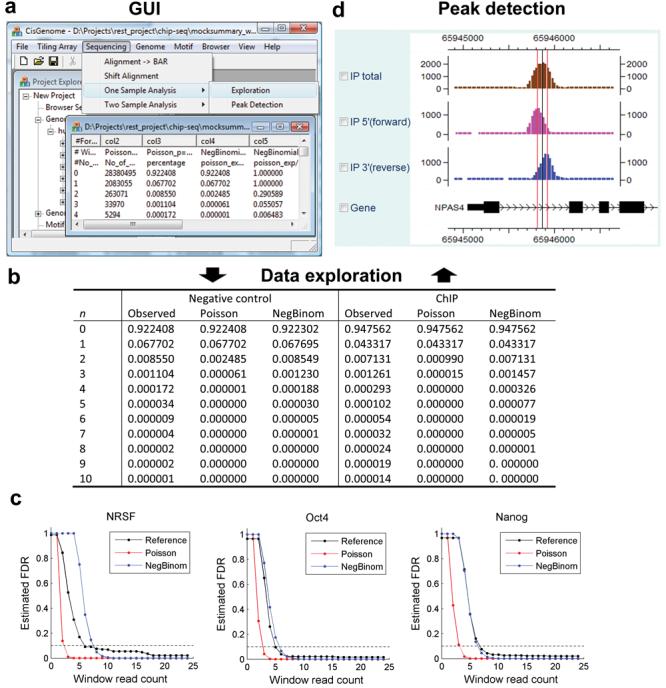
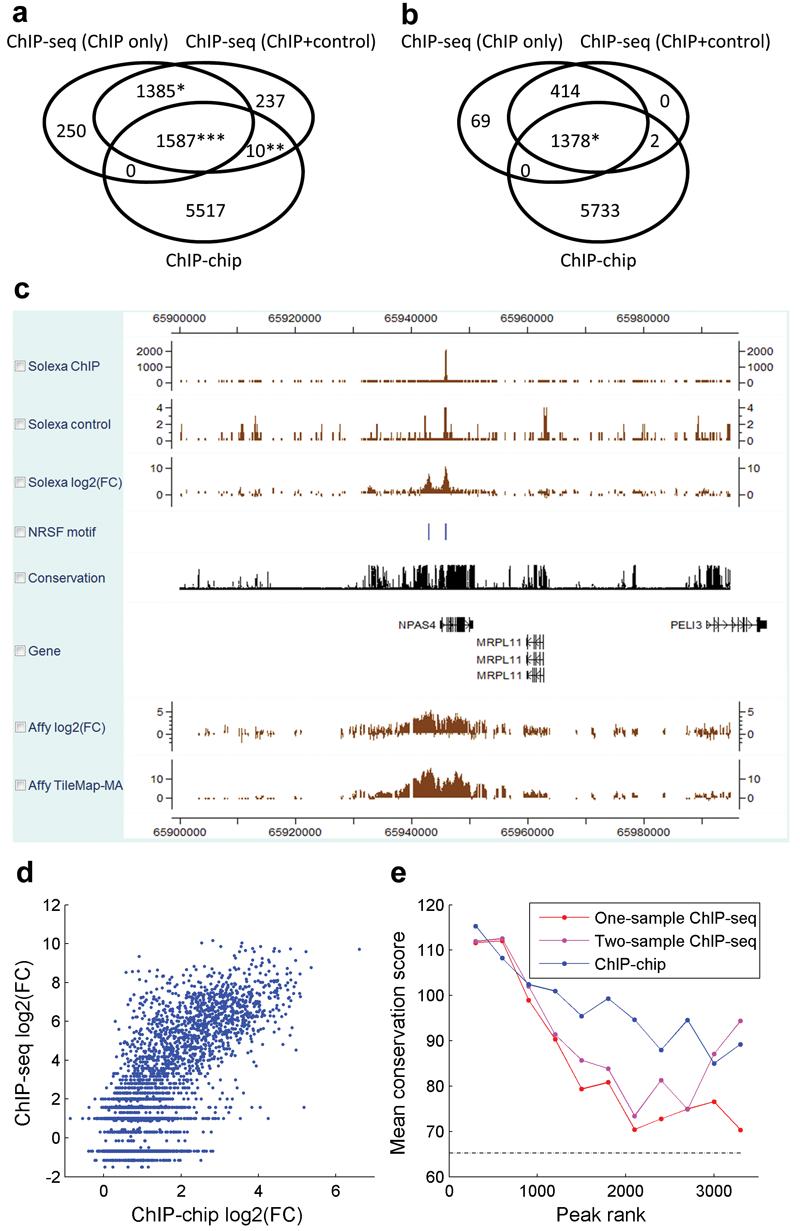
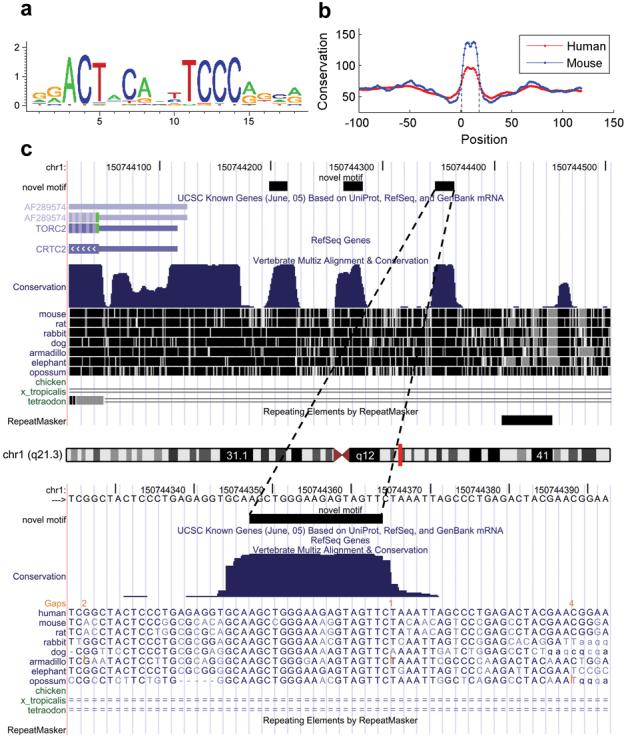
We present CisGenome, a software system for analyzing genome-wide chromatin immunoprecipitation (ChIP) data. CisGenome is designed to meet all basic needs of ChIP data analyses, including visualization, data normalization, peak detection, false discovery rate computation, gene-peak association, and sequence and motif analysis. In addition to implementing previously published ChIP-microarray (ChIP-chip) analysis methods, the software contains statistical methods designed specifically for ChlP sequencing (ChIP-seq) data obtained by coupling ChIP with massively parallel sequencing. The modular design of CisGenome enables it to support interactive analyses through a graphic user interface as well as customized batch-mode computation for advanced data mining. A built-in browser allows visualization of array images, signals, gene structure, conservation, and DNA sequence and motif information. We demonstrate the use of these tools by a comparative analysis of ChIP-chip and ChIP-seq data for the transcription factor NRSF/REST, a study of ChIP-seq analysis with or without a negative control sample, and an analysis of a new motif in Nanog- and Sox2-binding regions.
Figures
References
-
- Cawley S, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell. 2004;116:499–509. - PubMed
-
- Carroll JS, et al. Genome-wide analysis of estrogen receptor binding sites. Nat. Genet. 2006;38:1289–1297. - PubMed
-
- Johnson DS, Mortazavi A, Myers RM, Wold B. Genome-wide mapping of in vivo protein-DNA interactions. Science. 2007;316:1497–1502. - PubMed
-
- Robertson G, et al. Genome-wide profiles of STAT1 DNA association using chromatin immunoprecipitation and massively parallel sequencing. Nat. Methods. 2007;4:651–657. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
