

Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP)
- PMID: 18979151
- PMCID: PMC3620015
- DOI: 10.1007/s00774-008-0879-8
Skeletal metamorphosis in fibrodysplasia ossificans progressiva (FOP)
Abstract
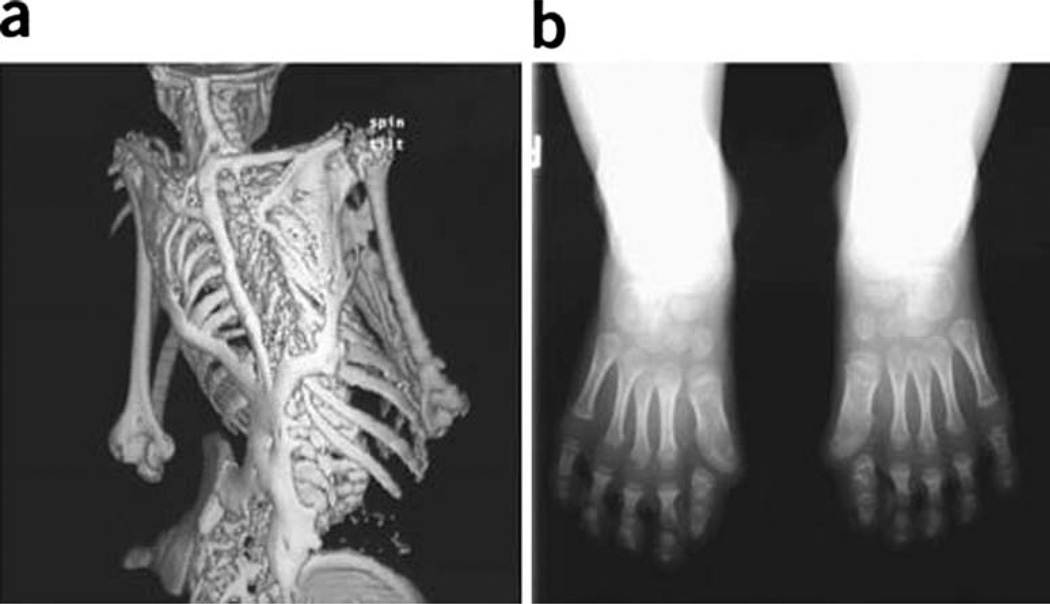
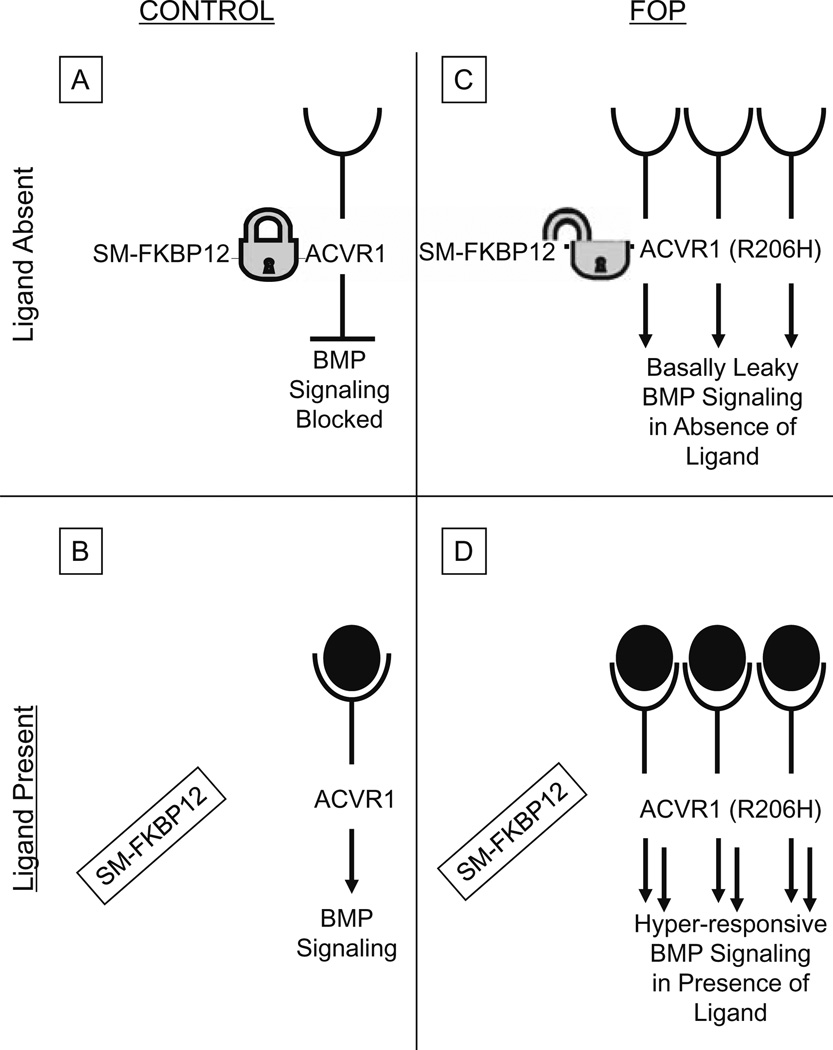
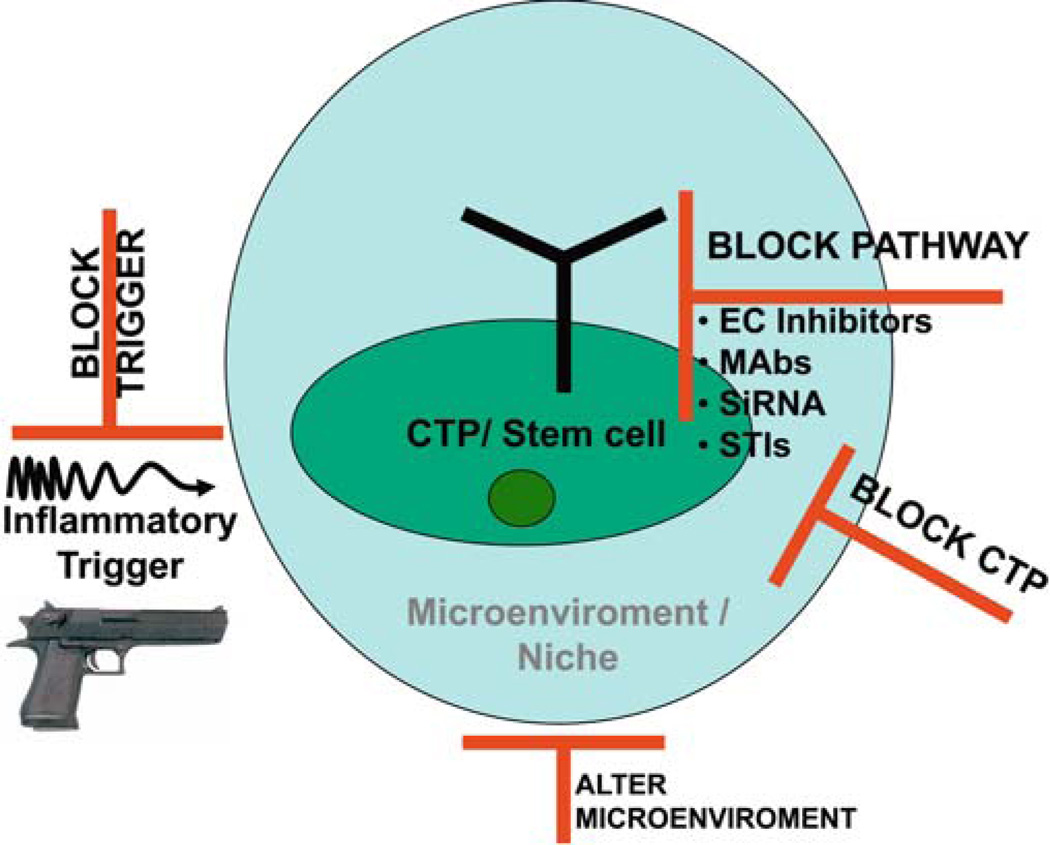
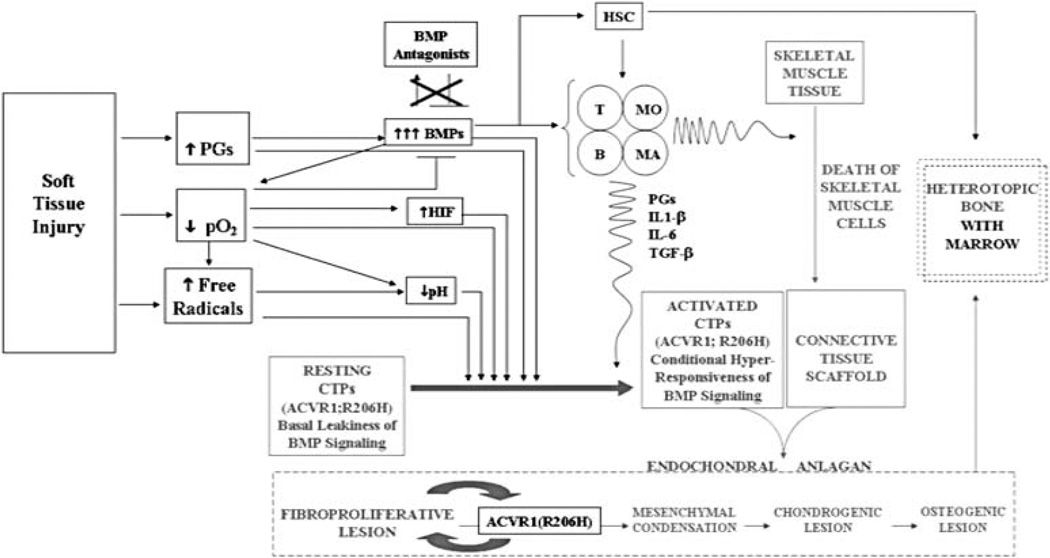
Metamorphosis, the transformation of one normal tissue or organ system into another, is a biological process rarely studied in higher vertebrates or mammals, but exemplified pathologically by the extremely disabling autosomal dominant disorder fibrodysplasia ossificans progressiva (FOP). The recurrent single nucleotide missense mutation in the gene encoding activin receptor IA/activin-like kinase-2 (ACVR1/ALK2), a bone morphogenetic protein type I receptor that causes skeletal metamorphosis in all classically affected individuals worldwide, is the first identified human metamorphogene. Physiological studies of this metamorphogene are beginning to provide deep insight into a highly conserved signaling pathway that regulates tissue stability following morphogenesis, and that when damaged at a highly specific locus (c.617G > A; R206H), and triggered by an inflammatory stimulus permits the renegade metamorphosis of normal functioning connective tissue into a highly ramified skeleton of heterotopic bone. A comprehensive understanding of the process of skeletal metamorphosis, as revealed by the rare condition FOP, will lead to the development of more effective treatments for FOP and, possibly, for more common disorders of skeletal metamorphosis.
Figures
References
-
- Kaplan FS, Glaser DL, Pignolo RJ, Shore EM. Introduction. Clin Rev Bone Miner Metab. 2005;3(3–4):175–177.
-
- Kaplan FS, Glaser DL, Shore EM, Deirmengian GK, Gupta R, Delai P, Morhart R, Smith R, Le Merrer M, Rogers JG, Connor JM, Kitterman JA. The phenotype of fibrodysplasia ossificans progressiva. Clin Rev Bone Miner Metab. 2005;3:183–188.
-
- Shore EM, Xu M, Feldman GJ, Fenstermacher DA, Cho T-J, Choi IH, Connor JM, Delai P, Glaser DL, Le Merrer M, Morhart R, Rogers JG, Smith R, Triffitt JT, Urtizberea JA, Zasloff M, Brown MA, Kaplan FS. A recurrent mutation in the BMP type I receptor ACVR1 causes inherited and sporadic fibrodysplasia ossificans progressiva. Nat Genet. 2006;38:525–527. - PubMed
-
- Cohen RB, Hahn GV, Tabas J, Peeper J, Levitz CL, Sando A, Sando N, Zasloff M, Kaplan FS. The natural history of heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. J Bone Joint Surg Am. 1993;75:215–219. - PubMed
-
- Rocke DM, Zasloff M, Peeper J, Cohen RB, Kaplan FS. Age and joint-specific risk of initial heterotopic ossification in patients who have fibrodysplasia ossificans progressiva. Clin Orthop Relat Res. 1994;301:243–248. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
