

Impeded electron transfer from a pathogenic FMN domain mutant of methionine synthase reductase and its responsiveness to flavin supplementation
- PMID: 18980384
- PMCID: PMC2645915
- DOI: 10.1021/bi8008328
Impeded electron transfer from a pathogenic FMN domain mutant of methionine synthase reductase and its responsiveness to flavin supplementation
Abstract
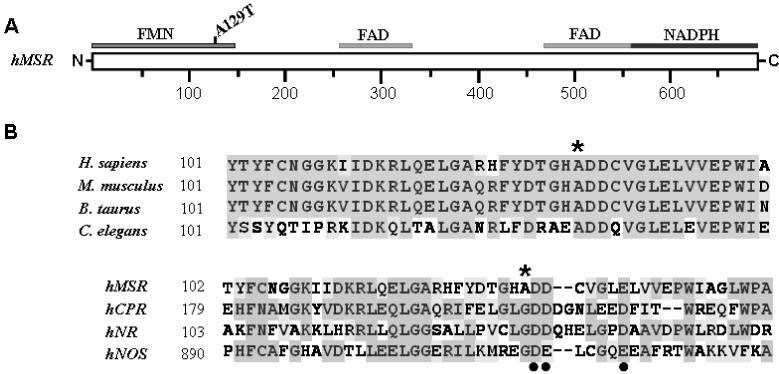
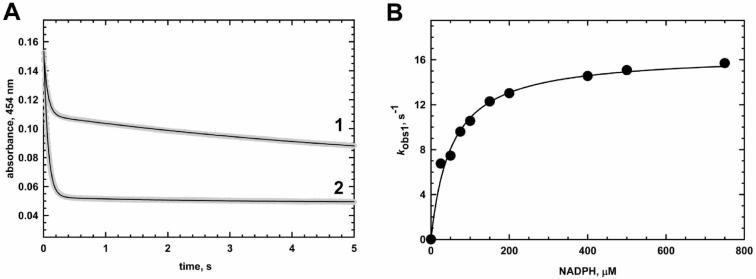
Methionine synthase reductase (MSR) is a diflavin oxidoreductase that transfers electrons from NADPH to oxidized cobalamin and plays a vital role in repairing inactive cobalamin-dependent methionine synthase. MSR deficiency is a recessive genetic disorder affecting folate and methionine metabolism and is characterized by elevated levels of plasma homocysteine. In this study, we have examined the molecular basis of MSR dysfunction associated with a patient mutation, A129T, which is housed in the FMN binding domain and is adjacent to a cluster of conserved acidic residues found in diflavin oxidoreductases. We show that the substitution of alanine with threonine destabilizes FMN binding without affecting the NADPH coenzyme specificity or affinity, indicating that the mutation's effects may be confined to the FMN module. The A129T MSR mutant transfers electrons to ferricyanide as efficiently as wild type MSR but the rate of cytochrome c, 2,6-dichloroindophenol, and menadione reduction is decreased 10-15 fold. The mutant is depleted in FMN and reactivates methionine synthase with 8% of the efficiency of wild type MSR. Reconstitution of A129T MSR with FMN partially restores its ability to reduce cytochrome c and to reactivate methionine synthase. Hydrogen-deuterium exchange mass spectrometric studies localize changes in backbone amide exchange rates to peptides in the FMN-binding domain. Together, our results reveal that the primary biochemical penalty associated with the A129T MSR mutant is its lower FMN content, provide insights into the distinct roles of the FAD and FMN centers in human MSR for delivering electrons to various electron acceptors, and suggest that patients harboring the A129T mutation may be responsive to riboflavin therapy.
Figures




References
-
- Mills JL, McPartlin JM, Kirke PN, Lee YJ, Conley MR, Weir DG, Scott JM. Homocysteine metabolism in pregnancies complicated by neural-tube defects. Lancet. 1995;345:149–151. - PubMed
-
- Clarke R, Smith AD, Jobst KA, Refsum H, Sutton L, Ueland PM. Folate, vitamin B12, and serum total homocysteine levels in confirmed Alzheimer disease. Arch Neurol. 1998;55:1449–1455. - PubMed
-
- Refsum H, Ueland PM, Nygard O, Vollset SE. Homocysteine and Cardiovascular Disease. Annu. Rev. Medicine. 1998;49:31–62. - PubMed
-
- Rosenblatt DS. Inborn errors of folate and cobalamin metabolism. Cambridge University Press; Cambridge: 2001.
-
- Finkelstein JD. Inborn errors of sulfur-containing amino acid metabolism. J Nutr. 2006;136:1750S–1754S. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
