

Improving gemcitabine-mediated radiosensitization using molecularly targeted therapy: a review
- PMID: 18980967
- PMCID: PMC2697824
- DOI: 10.1158/1078-0432.CCR-08-1032
Improving gemcitabine-mediated radiosensitization using molecularly targeted therapy: a review
Abstract
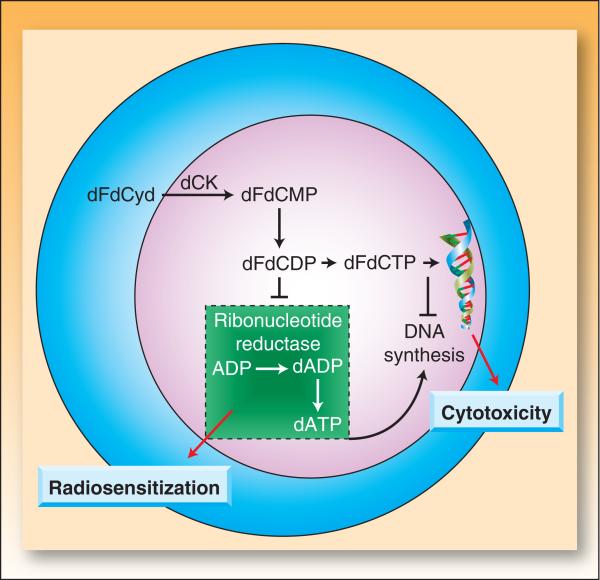
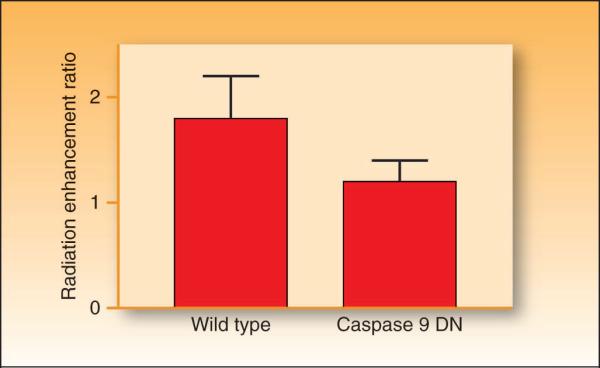
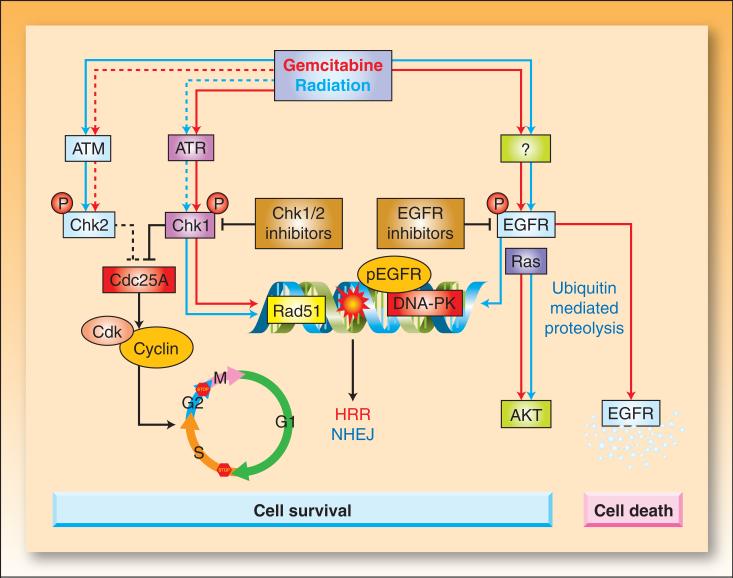
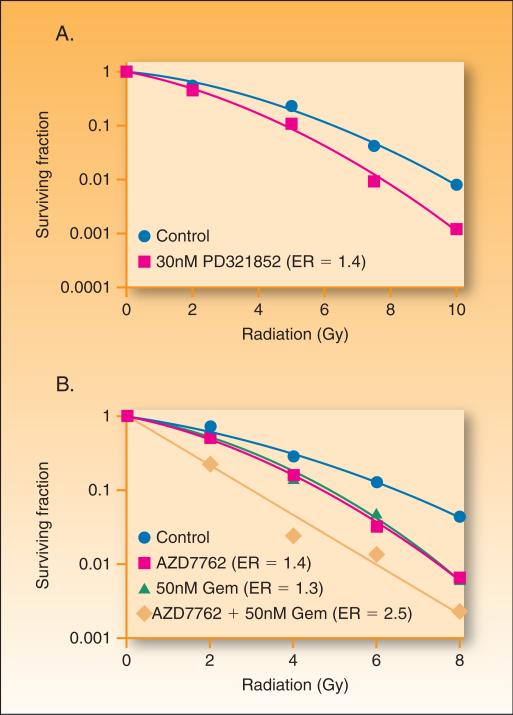
In the last three decades, gemcitabine has progressed from the status of a laboratory cytotoxic drug to a standard clinical chemotherapeutic agent and a potent radiation sensitizer. In an effort to improve the efficacy of gemcitabine, additional chemotherapeutic agents have been combined with gemcitabine (both with and without radiation) but with toxicity proving to be a major limitation. Therefore, the integration of molecularly targeted agents, which potentially produce less toxicity than standard chemotherapy, with gemcitabine radiation is a promising strategy for improving chemoradiation. Two of the most promising targets, described in this review, for improving the efficacy of gemcitabine radiation are epidermal growth factor receptor and checkpoint kinase 1.
Figures
References
-
- Safran H, Dipetrillo T, Iannitti D, et al. Gemcitabine, paclitaxel, and radiation for locally advanced pancreatic cancer: a Phase I trial. International journal of radiation oncology, biology, physics. 2002;54:137–41. - PubMed
-
- Blackstock AW, Melin SA, Butler JM, et al. Irinotecan/gemcitabine followed by twice-weekly gemcitabine/radiation in locally advanced pancreatic cancer. Oncology (Williston Park) 2002;16:25–8. - PubMed
-
- Kachnic LA, Shaw JE, Manning MA, Lauve AD, Neifeld JP. Gemcitabine following radiotherapy with concurrent 5-fluorouracil for nonmetastatic adenocarcinoma of the pancreas. International journal of cancer. 2001;96:132–9. - PubMed
-
- Symon Z, Davis M, McGinn CJ, Zalupski MM, Lawrence TS. Concurrent chemoradiotherapy with gemcitabine and cisplatin for pancreatic cancer: from the laboratory to the clinic. International journal of radiation oncology, biology, physics. 2002;53:140–5. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
