

Inhibitory control over Ca(2+) sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression
- PMID: 18982068
- PMCID: PMC2575405
- DOI: 10.1371/journal.pone.0003644
Inhibitory control over Ca(2+) sparks via mechanosensitive channels is disrupted in dystrophin deficient muscle but restored by mini-dystrophin expression
Abstract
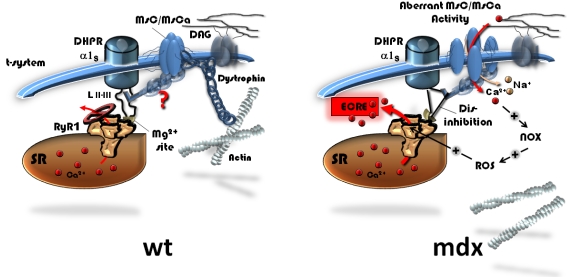
Background: In dystrophic skeletal muscle, osmotic stimuli somehow relieve inhibitory control of dihydropyridine receptors (DHPR) on spontaneous sarcoplasmic reticulum elementary Ca(2+) release events (ECRE) in high Ca(2+) external environments. Such 'uncontrolled' Ca(2+) sparks were suggested to act as dystrophic signals. They may be related to mechanosensitive pathways but the mechanisms are elusive. Also, it is not known whether truncated dystrophins can correct the dystrophic disinhibition.
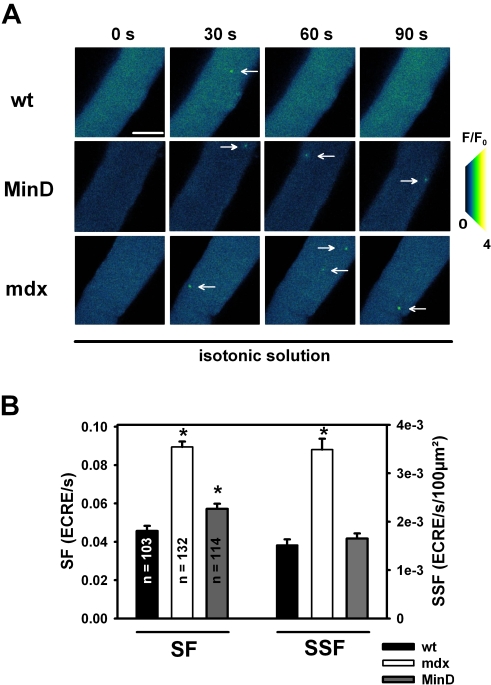
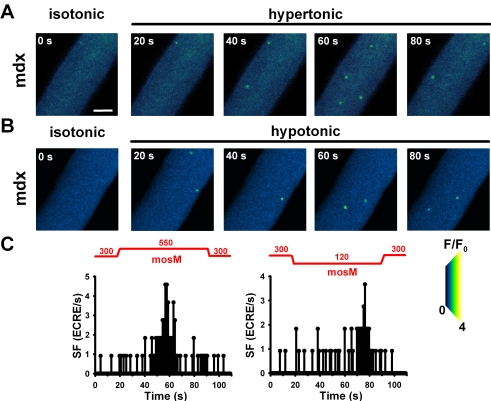
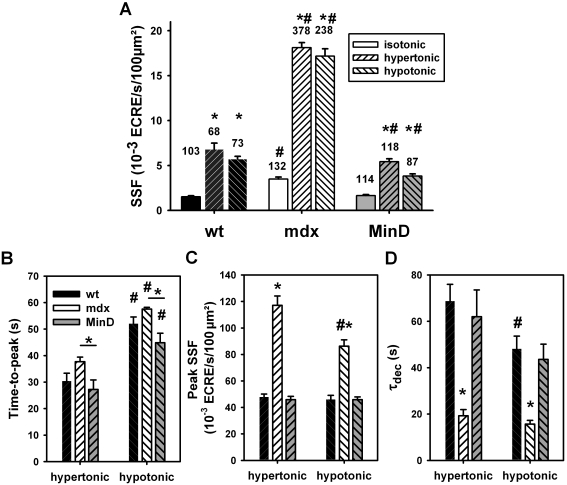
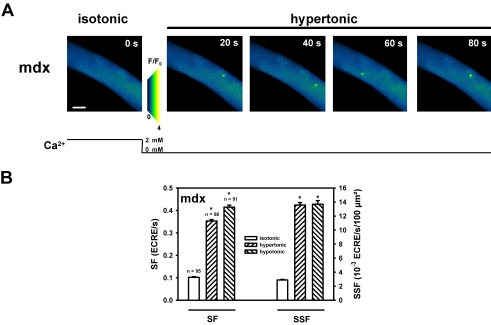
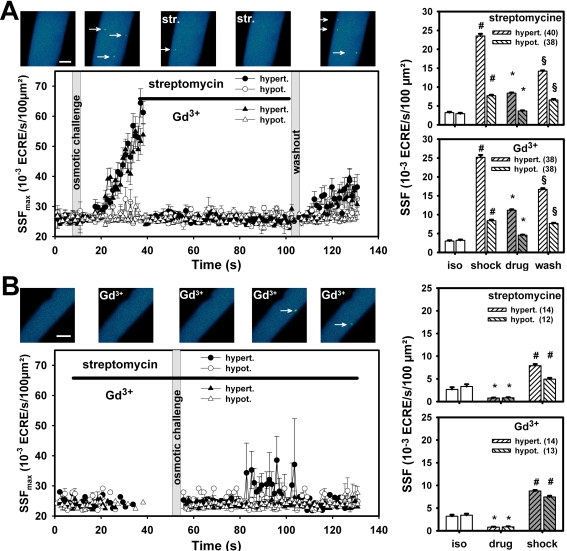
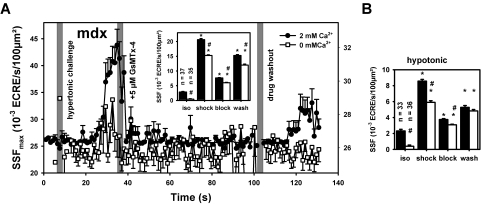
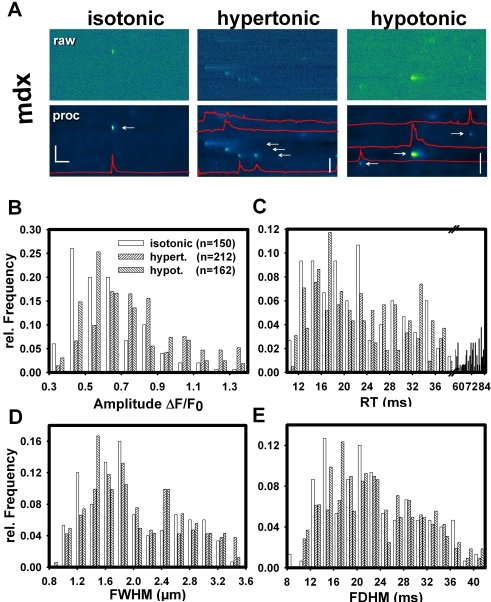
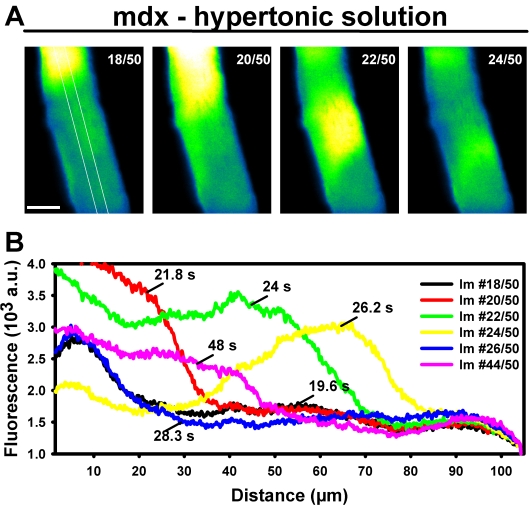
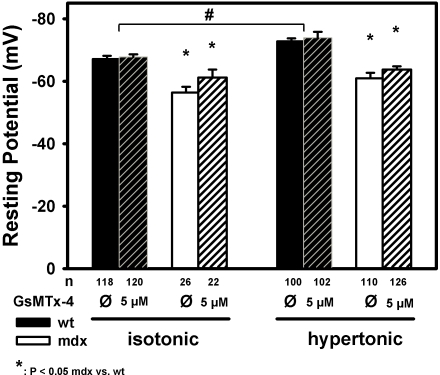
Methodology/principal findings: We recorded ECRE activity in single intact fibers from adult wt, mdx and mini-dystrophin expressing mice (MinD) under resting isotonic conditions and following hyper-/hypo-osmolar external shock using confocal microscopy and imaging techniques. Isotonic ECRE frequencies were small in wt and MinD fibers, but were markedly increased in mdx fibers. Osmotic challenge dramatically increased ECRE activity in mdx fibers. Sustained osmotic challenge induced marked exponential ECRE activity adaptation that was three times faster in mdx compared to wt and MinD fibers. Rising external Ca(2+) concentrations amplified osmotic ECRE responses. The eliminated ECRE suppression in intact osmotically stressed mdx fibers was completely and reversibly resuscitated by streptomycine (200 microM), spider peptide GsMTx-4 (5 microM) and Gd(3+) (20 microM) that block unspecific, specific cationic and Ca(2+) selective mechanosensitive channels (MsC), respectively. ECRE morphology was not substantially altered by membrane stress. During hyperosmotic challenge, membrane potentials were polarised and a putative depolarisation through aberrant MsC negligible excluding direct activation of ECRE through tubular depolarisation.
Conclusions/significance: Dystrophin suppresses spontaneous ECRE activity by control of mechanosensitive pathways which are suggested to interact with the inhibitory DHPR loop to the ryanodine receptor. MsC-related disinhibition prevails in dystrophic muscle and can be resuscitated by transgenic mini-dystrophin expression. Our results have important implications for the pathophysiology of DMD where abnormal MsC in dystrophic muscle confer disruption of microdomain Ca(2+) homeostasis. MsC blockers should have considerable therapeutic potential if more muscle specific compounds can be found.
Conflict of interest statement
Figures
Similar articles
-
L-type Ca2+ channel function is linked to dystrophin expression in mammalian muscle.PLoS One. 2008 Mar 12;3(3):e1762. doi: 10.1371/journal.pone.0001762. PLoS One. 2008. PMID: 18516256 Free PMC article.
-
Involvement of TRPC in the abnormal calcium influx observed in dystrophic (mdx) mouse skeletal muscle fibers.J Cell Biol. 2002 Sep 16;158(6):1089-96. doi: 10.1083/jcb.200203091. Epub 2002 Sep 16. J Cell Biol. 2002. PMID: 12235126 Free PMC article.
-
In situ measurements of calpain activity in isolated muscle fibres from normal and dystrophin-lacking mdx mice.J Physiol. 2007 Aug 1;582(Pt 3):1261-75. doi: 10.1113/jphysiol.2007.132191. Epub 2007 May 17. J Physiol. 2007. PMID: 17510188 Free PMC article.
-
Ca2+ sparks as a plastic signal for skeletal muscle health, aging, and dystrophy.Acta Pharmacol Sin. 2006 Jul;27(7):791-8. doi: 10.1111/j.1745-7254.2006.00384.x. Acta Pharmacol Sin. 2006. PMID: 16787561 Review.
-
Sarcolemmal ion channels in dystrophin-deficient skeletal muscle fibres.J Muscle Res Cell Motil. 2006;27(5-7):367-73. doi: 10.1007/s10974-006-9083-4. Epub 2006 Jul 28. J Muscle Res Cell Motil. 2006. PMID: 16874448 Review.
Cited by
-
Comparative proteomic profiling of soleus, extensor digitorum longus, flexor digitorum brevis and interosseus muscles from the mdx mouse model of Duchenne muscular dystrophy.Int J Mol Med. 2013 Sep;32(3):544-56. doi: 10.3892/ijmm.2013.1429. Epub 2013 Jul 3. Int J Mol Med. 2013. PMID: 23828267 Free PMC article.
-
GsMTx4-D provides protection to the D2.mdx mouse.Neuromuscul Disord. 2018 Oct;28(10):868-877. doi: 10.1016/j.nmd.2018.07.005. Epub 2018 Jul 21. Neuromuscul Disord. 2018. PMID: 30174173 Free PMC article.
-
Myofibrillar misalignment correlated to triad disappearance of mdx mouse gastrocnemius muscle probed by SHG microscopy.Biomed Opt Express. 2014 Feb 25;5(3):858-75. doi: 10.1364/BOE.5.000858. eCollection 2014 Mar 1. Biomed Opt Express. 2014. PMID: 24688819 Free PMC article.
-
Current Pharmacological Strategies for Duchenne Muscular Dystrophy.Front Cell Dev Biol. 2021 Aug 19;9:689533. doi: 10.3389/fcell.2021.689533. eCollection 2021. Front Cell Dev Biol. 2021. PMID: 34490244 Free PMC article. Review.
-
New factors contributing to dynamic calcium regulation in the skeletal muscle triad-a crowded place.Biophys Rev. 2010 Feb;2(1):29-38. doi: 10.1007/s12551-009-0027-2. Epub 2009 Dec 18. Biophys Rev. 2010. PMID: 28509943 Free PMC article. Review.
References
-
- Hoffman EP, Brown RH, Kunkel LM. Dystrophin: the protein product of the Duchenne muscular dystrophy locus. Cell. 1987;51:919–928. - PubMed
-
- Campbell KP. Three muscular dystrophies: loss of cytoskeleton-extracellular matrix linkage. Cell. 1995;80:675–679. - PubMed
-
- Minetti GC, Colussi C, Adami R, Serra C, Mozzetta C, et al. Functional and morphological recovery of dystrophic muscles in mice treated with deacetylase inhibitors. Nat Med. 2006;12(10):1147–1150. - PubMed
-
- De Luca A, Pierno S, Liantonio A, Cetrone M, Camerino C, et al. Enhanced dystrophic progression in mdx mice by exercise and beneficial effects of taurine and insulin-like growth factor-1. J Pharmacol Exp Ther. 2003;304(1):453–463. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
