

Kallmann syndrome
- PMID: 18985070
- PMCID: PMC2986064
- DOI: 10.1038/ejhg.2008.206
Kallmann syndrome
Abstract
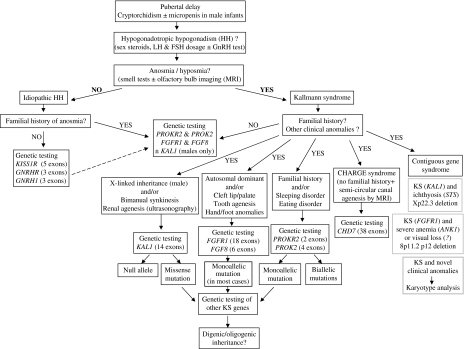
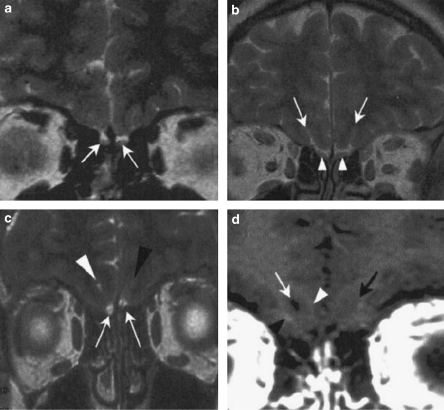
The Kallmann syndrome (KS) combines hypogonadotropic hypogonadism (HH) with anosmia. This is a clinically and genetically heterogeneous disease. KAL1, encoding the extracellular glycoprotein anosmin-1, is responsible for the X chromosome-linked recessive form of the disease. Mutations in FGFR1 or FGF8, encoding fibroblast growth factor receptor-1 and fibroblast growth factor-8, respectively, underlie an autosomal dominant form with incomplete penetrance. Finally, mutations in PROKR2 and PROK2, encoding prokineticin receptor-2 and prokineticin-2, have been found in heterozygous, homozygous, and compound heterozygous states. These two genes are likely to be involved both in monogenic recessive and digenic/oligogenic KS transmission modes. Notably, mutations in any of the above-mentioned KS genes have been found in less than 30% of the KS patients, which indicates that other genes involved in the disease remain to be discovered.
Figures
References
-
- Maestre de San Juan A. Falta total de los nervios olfactorios con anosmía en un individuo en quien existia una atrofía congénita de los testículos y miembro viril. Siglo Medico. 1856;131:211.
-
- Kallmann FJ, Schoenfeld WA, Barrera SE. The genetic aspects of primary eunuchoidism. Am J Mental Deficiency. 1944;XLVIII:203–236.
-
- de Morsier G, Gauthier G. La dysplasie olfacto-génitale. Pathol Biol. 1963;11:1267–1272. - PubMed
-
- Naftolin F, Harris GW, Bobrow M. Effect of purified luteinizing hormone releasing factor on normal and hypogonadotropic anosmic men. Nature. 1971;232:496–497. - PubMed
-
- Santen RJ, Paulsen CA. Hypogonadotropic eunuchoidism. I. Clinical study of the mode of inheritance. J Clin Endocrinol Metab. 1972;36:47–54. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
