

Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator
- PMID: 18990670
- PMCID: PMC2674095
- DOI: 10.1098/rstb.2008.0142
Review. SUR1: a unique ATP-binding cassette protein that functions as an ion channel regulator
Abstract
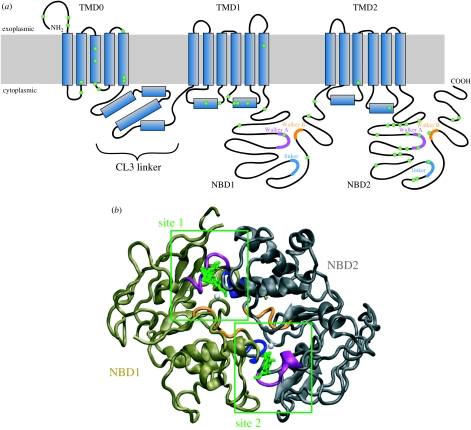
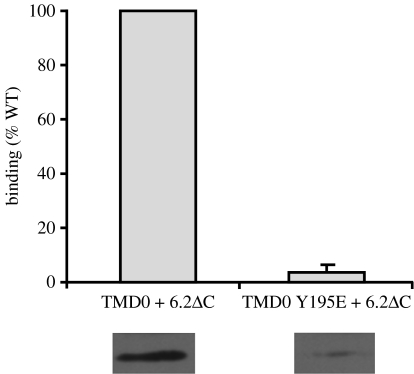
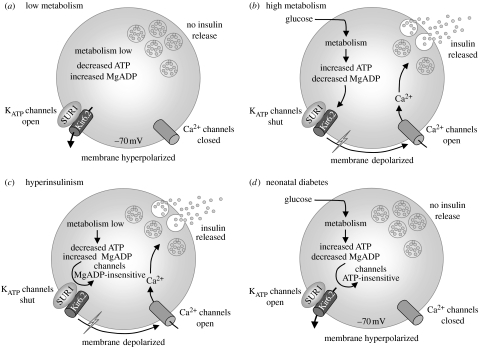
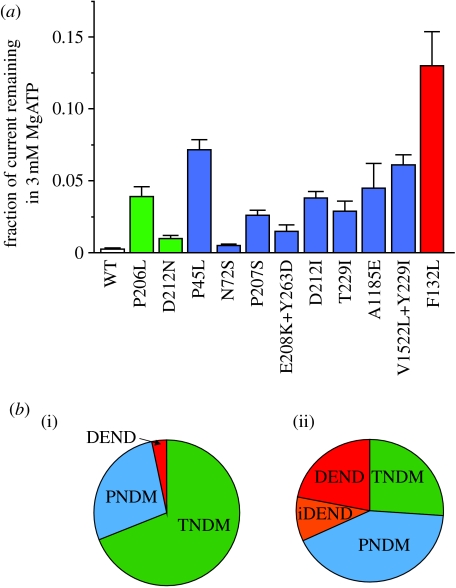
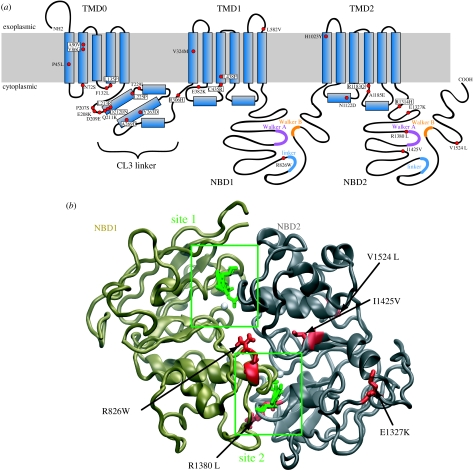
SUR1 is an ATP-binding cassette (ABC) transporter with a novel function. In contrast to other ABC proteins, it serves as the regulatory subunit of an ion channel. The ATP-sensitive (KATP) channel is an octameric complex of four pore-forming Kir6.2 subunits and four regulatory SUR1 subunits, and it links cell metabolism to electrical activity in many cell types. ATPase activity at the nucleotide-binding domains of SUR results in an increase in KATP channel open probability. Conversely, ATP binding to Kir6.2 closes the channel. Metabolic regulation is achieved by the balance between these two opposing effects. Precisely how SUR1 talks to Kir6.2 remains unclear, but recent studies have identified some residues and domains that are involved in both physical and functional interactions between the two proteins. The importance of these interactions is exemplified by the fact that impaired regulation of Kir6.2 by SUR1 results in human disease, with loss-of-function SUR1 mutations causing congenital hyperinsulinism and gain-of-function SUR1 mutations leading to neonatal diabetes. This paper reviews recent data on the regulation of Kir6.2 by SUR1 and considers the molecular mechanisms by which SUR1 mutations produce disease.
Figures
References
-
- Abdulhadi-Atwan M., Bushman J.D., Tornovsky-Babaey S., Perry A., Abu-Libdeh A., Glaser B., Shyng S.L., Zangen D.H. Novel de novo mutation in SUR1 presenting as HI in infancy followed by overt diabetes in early adolescence. Diabetes. 2008;57:1935–1940. doi:10.2337/db08-0159 - DOI - PMC - PubMed
-
- Ashcroft F.M. ATP-sensitive potassium channelopathies: focus on insulin secretion. J. Clin. Invest. 2005;115:2047–2058. doi:10.1172/JCI25495 - DOI - PMC - PubMed
-
- Ashcroft F.M. The Walter B. cannon physiology in perspective lecture, 2007 ATP-sensitive K+ channels and disease: from molecule to malady. Am. J. Physiol. Endocrinol. Metab. 2007;293:E880–E889. doi:10.1152/ajpendo.00348.2007 - DOI - PubMed
-
- Ashfield R., Gribble F.M., Ashcroft S.J., Ashcroft F.M. Identification of the high-affinity tolbutamide site on the SUR1 subunit of the KATP channel. Diabetes. 1999;48:1341–1347. doi:10.2337/diabetes.48.6.1341 - DOI - PubMed
-
- Babenko A.P. A novel ABCC8 (SUR1)-dependent mechanism of metabolism-excitation uncoupling. J. Biol. Chem. 2008;283:8778–8782. doi:10.1074/jbc.C700243200 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources