

Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders
- PMID: 18991648
- PMCID: PMC3230043
- DOI: 10.2174/156652408786241447
Nitric oxide and arginine dysregulation: a novel pathway to pulmonary hypertension in hemolytic disorders
Abstract
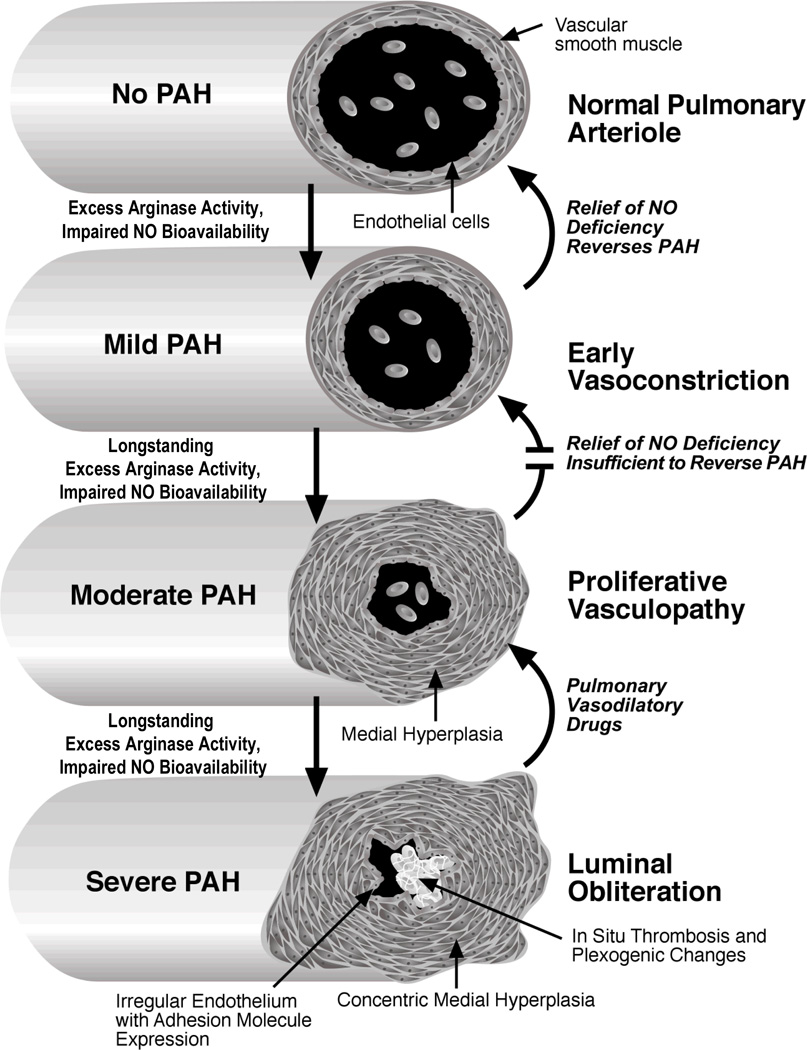
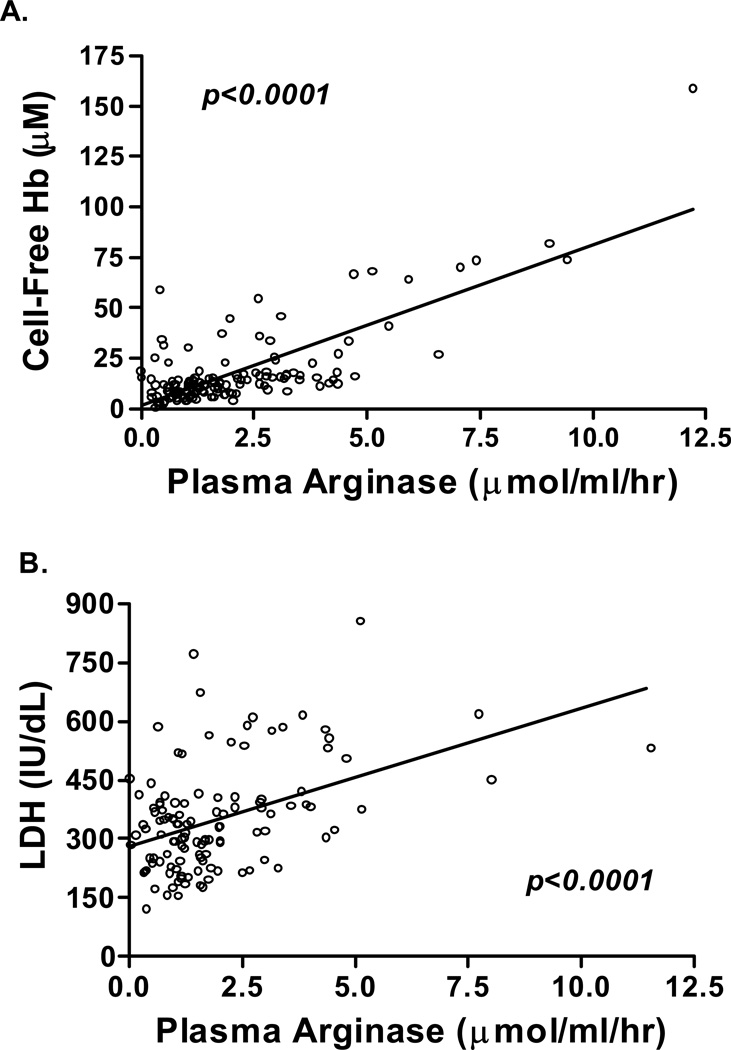
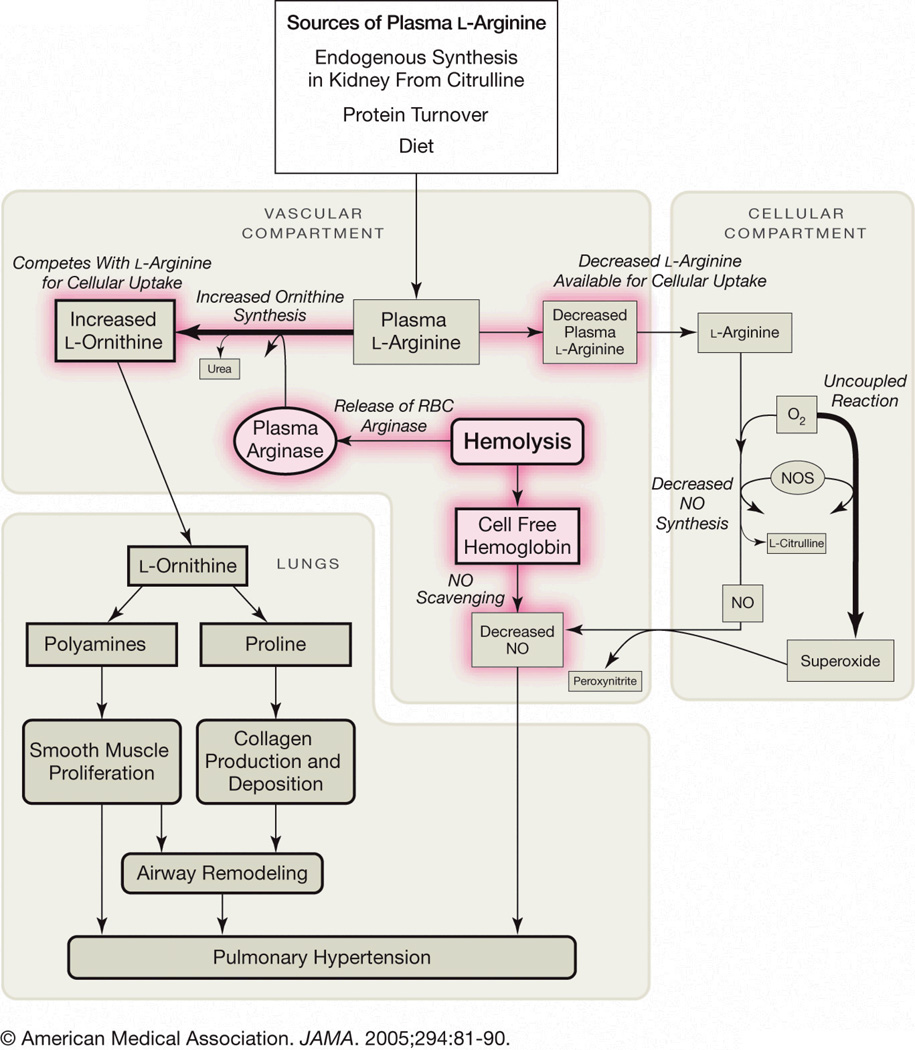
Secondary pulmonary hypertension (PH) is emerging as one of the leading causes of mortality and morbidity in patients with hemolytic anemias such as sickle cell disease (SCD) and thalassemia. Impaired nitric oxide (NO) bioavailability represents the central feature of endothelial dysfunction, and is a major factor in the pathophysiology of PH. Inactivation of NO correlates with hemolytic rate and is associated with the erythrocyte release of cell-free hemoglobin, which consumes NO directly, and the simultaneous release of the arginine-metabolizing enzyme arginase, which limits bioavailability of the NO synthase substrate arginine during the process of intravascular hemolysis. Rapid consumption of NO is accelerated by oxygen radicals that exists in both SCD and thalassemia. A dysregulation of arginine metabolism contributes to endothelial dysfunction and PH in SCD, and is strongly associated with prospective patient mortality. The central mechanism responsible for this metabolic disorder is enhanced arginine turnover, occurring secondary to enhanced plasma arginase activity. This is consistent with a growing appreciation of the role of excessive arginase activity in human diseases, including asthma and pulmonary arterial hypertension. New treatments aimed at improving arginine and NO bioavailability through arginase inhibition, suppression of hemolytic rate, oral arginine supplementation, or use of NO donors represent potential therapeutic strategies for this common pulmonary complication of hemolytic disorders.
Conflict of interest statement
Authors declare no conflicts of interest.
Figures
References
-
- Bunn HF. Pathogenesis and treatment of sickle cell disease. N Engl J Med. 1997;337:762–769. - PubMed
-
- Stuart MJ, Nagel RL. Sickle-cell disease. Lancet. 2004;364:1343–1360. - PubMed
-
- Hebbel RP, Osarogiagbon KD. The endothelial biology of sickle cell disease: Inflammation and a chronic vasculopathy. Microcirculation. 2004;11:129–151. - PubMed
-
- Frenette PS. Sickle cell vaso-occlusion: multistep and multicellular paradigm. Curr Opin Hematol. 2002;9:101–106. - PubMed
-
- Parise LV, Telen MJ. Erythrocyte adhesion in sickle cell disease. Curr Hematol Rep. 2003;2:102–108. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical