

Analysis of the genetic variation in Mycobacterium tuberculosis strains by multiple genome alignments
- PMID: 18992142
- PMCID: PMC2590607
- DOI: 10.1186/1756-0500-1-110
Analysis of the genetic variation in Mycobacterium tuberculosis strains by multiple genome alignments
Abstract
Background: The recent determination of the complete nucleotide sequence of several Mycobacterium tuberculosis (MTB) genomes allows the use of comparative genomics as a tool for dissecting the nature and consequence of genetic variability within this species. The multiple alignment of the genomes of clinical strains (CDC1551, F11, Haarlem and C), along with the genomes of laboratory strains (H37Rv and H37Ra), provides new insights on the mechanisms of adaptation of this bacterium to the human host.
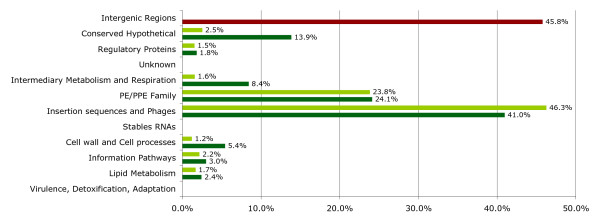
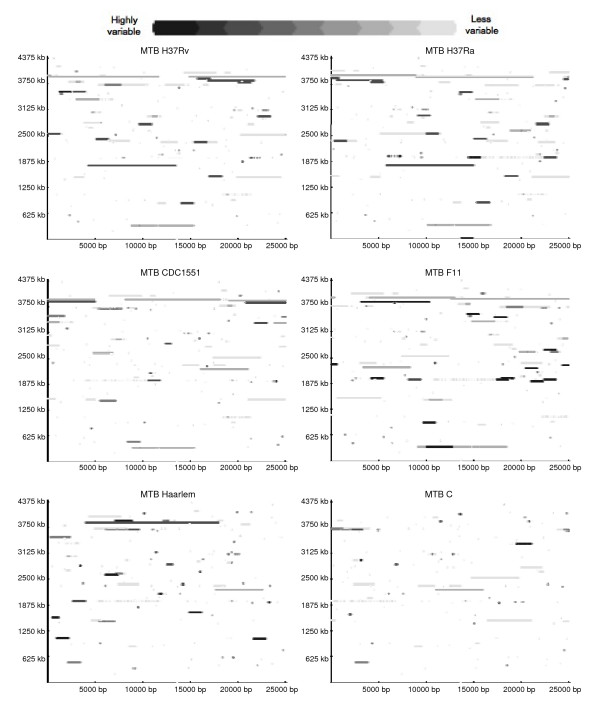
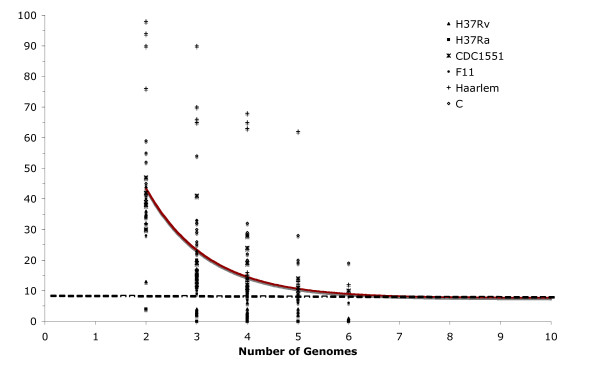
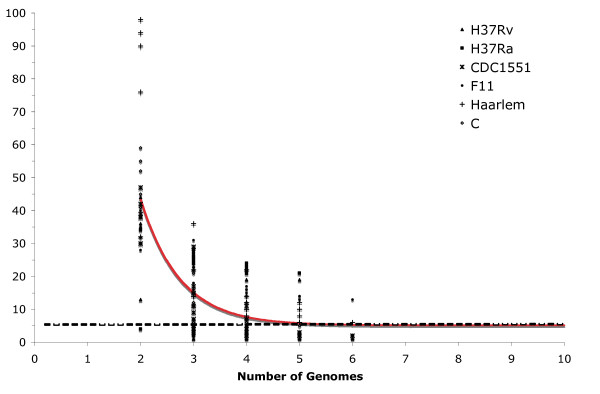
Findings: The genetic variation found in six M. tuberculosis strains does not involve significant genomic rearrangements. Most of the variation results from deletion and transposition events preferentially associated with insertion sequences and genes of the PE/PPE family but not with genes implicated in virulence. Using a Perl-based software islandsanalyser, which creates a representation of the genetic variation in the genome, we identified differences in the patterns of distribution and frequency of the polymorphisms across the genome. The identification of genes displaying strain-specific polymorphisms and the extrapolation of the number of strain-specific polymorphisms to an unlimited number of genomes indicates that the different strains contain a limited number of unique polymorphisms.
Conclusion: The comparison of multiple genomes demonstrates that the M. tuberculosis genome is currently undergoing an active process of gene decay, analogous to the adaptation process of obligate bacterial symbionts. This observation opens new perspectives into the evolution and the understanding of the pathogenesis of this bacterium.
Figures
References
LinkOut - more resources
Full Text Sources
Miscellaneous
