

Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration
- PMID: 18996918
- PMCID: PMC2638803
- DOI: 10.1093/hmg/ddn375
Variants of the elongator protein 3 (ELP3) gene are associated with motor neuron degeneration
Abstract
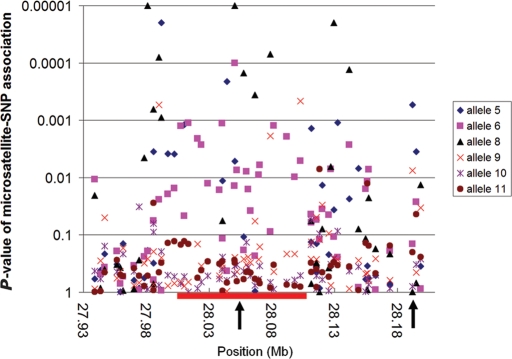
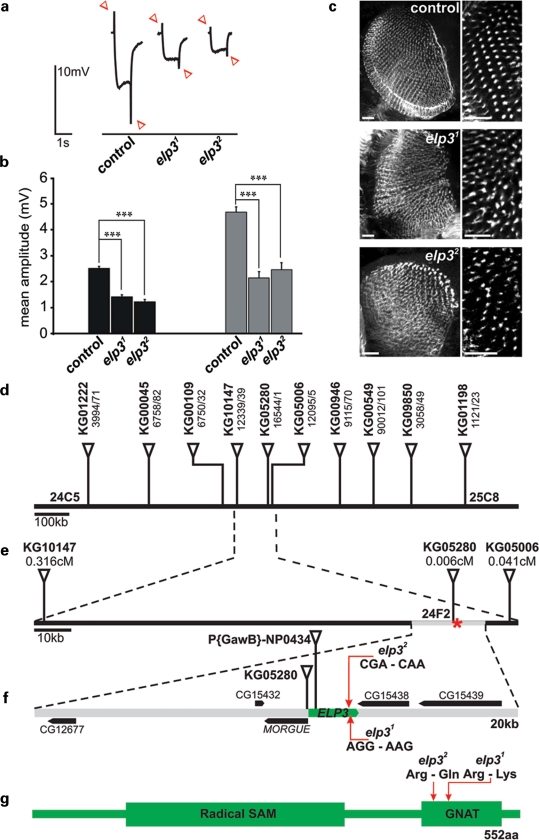
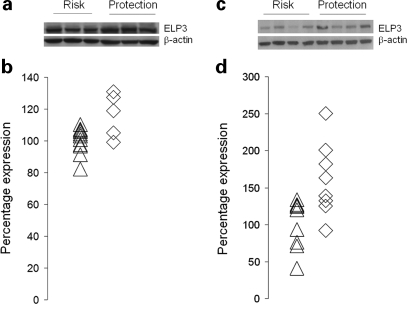
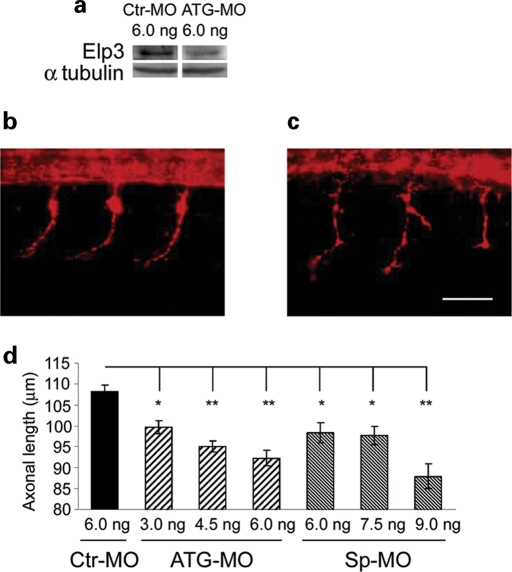
Amyotrophic lateral sclerosis (ALS) is a spontaneous, relentlessly progressive motor neuron disease, usually resulting in death from respiratory failure within 3 years. Variation in the genes SOD1 and TARDBP accounts for a small percentage of cases, and other genes have shown association in both candidate gene and genome-wide studies, but the genetic causes remain largely unknown. We have performed two independent parallel studies, both implicating the RNA polymerase II component, ELP3, in axonal biology and neuronal degeneration. In the first, an association study of 1884 microsatellite markers, allelic variants of ELP3 were associated with ALS in three human populations comprising 1483 people (P=1.96 x 10(-9)). In the second, an independent mutagenesis screen in Drosophila for genes important in neuronal communication and survival identified two different loss of function mutations, both in ELP3 (R475K and R456K). Furthermore, knock down of ELP3 protein levels using antisense morpholinos in zebrafish embryos resulted in dose-dependent motor axonal abnormalities [Pearson correlation: -0.49, P=1.83 x 10(-12) (start codon morpholino) and -0.46, P=4.05 x 10(-9) (splice-site morpholino), and in humans, risk-associated ELP3 genotypes correlated with reduced brain ELP3 expression (P=0.01). These findings add to the growing body of evidence implicating the RNA processing pathway in neurodegeneration and suggest a critical role for ELP3 in neuron biology and of ELP3 variants in ALS.
Figures
References
-
- Simpson C.L., Al-Chalabi A. Amyotrophic lateral sclerosis as a complex genetic disease. Biochim. Biophys. Acta. 2006;1762:973–985. - PubMed
-
- Kabashi E., Valdmanis P.N., Dion P., Spiegelman D., McConkey B.J., Velde C.V., Bouchard J.P., Lacomblez L., Pochigaeva K., Salachas F., et al. TARDBP mutations in individuals with sporadic and familial amyotrophic lateral sclerosis. Nat. Genet. 2008;40:572–574. - PubMed
-
- Schymick J.C., Scholz S.W., Fung H.C., Britton A., Arepalli S., Gibbs J.R., Lombardo F., Matarin M., Kasperaviciute D., Hernandez D.G., et al. Genome-wide genotyping in amyotrophic lateral sclerosis and neurologically normal controls: first stage analysis and public release of data. Lancet Neurol. 2007;6:322–328. - PubMed
-
- Dunckley T., Huentelman M.J., Craig D.W., Pearson J.V., Szelinger S., Joshipura K., Halperin R.F., Stamper C., Jensen K.R., Letizia D., et al. Whole-genome analysis of sporadic amyotrophic lateral sclerosis. N. Engl. J. Med. 2007;357:775–788. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
