

Genome pool strategy for structural coverage of protein families
- PMID: 19000818
- PMCID: PMC2902364
- DOI: 10.1016/j.str.2008.08.018
Genome pool strategy for structural coverage of protein families
Abstract
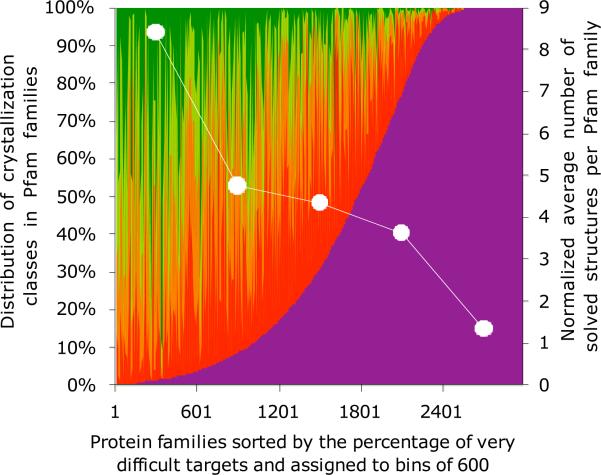
Even closely homologous proteins often have different crystallization properties and propensities. This observation can be used to introduce an additional dimension into crystallization trials by simultaneous targeting multiple homologs in what we call a "genome pool" strategy. We show that this strategy works because protein physicochemical properties correlated with crystallization success have a surprisingly broad distribution within most protein families. There are also "easy" and "difficult" families where this distribution is tilted in one direction. This leads to uneven structural coverage of protein families, with more "easy" ones solved. Increasing the size of the "genome pool" can improve chances of solving the "difficult" ones. In contrast, our analysis does not indicate that any specific genomes are "easy" or "difficult". Finally, we show that the group of proteins with known 3D structures is systematically different from the general pool of known proteins and we assess the structural consequences of these differences.
Figures




References
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. Basic local alignment search tool. J Mol Biol. 1990;215:403–410. - PubMed
-
- Carter CW, Jr., Carter CW. Protein crystallization using incomplete factorial experiments. J Biol Chem. 1979;254:12219–12223. - PubMed
-
- Chen L, Oughtred R, Berman HM, Westbrook J. TargetDB: a target registration database for structural genomics projects. Bioinformatics. 2004;20:2860–2862. - PubMed
-
- Creighton TE. Proteins: Structures and Molecular Properties. New York: 1984.
-
- Eddy SR. Profile hidden Markov models. Bioinformatics. 1998;14:755–763. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
